
Profil clinique, épidémiologique, thérapeutique et évolutif de la maladie de Vogt-Koyanagi-Harada: étude rétrospective d´une série de 10 cas au Maroc
Zinab Bott, Kadidiata Hamed Toure, Amal El-Ouakhoumi, Jaouad Yousfi, Laila Benjilali, Mouna Zahlane, Lamiaa Essaadouni
Corresponding author: Kadidiata Hamed Toure, Service de Médecine Interne, CHU Bouaké, Bouaké, Cote d´Ivoire 
Received: 08 Jun 2026 - Accepted: 15 Jun 2026 - Published: 10 Jul 2026
Domain: Internal medicine
Keywords: Syndrome de Vogt-Koyanagi-Harada, panuvéite, cyclophosphamide, agents immunosuppresseurs, acuité visuelle
Funding: Ce travail n'a reçu aucune subvention spécifique de la part d'organismes de financement du secteur public, commercial ou à but non lucratif.
©Zinab Bott et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Zinab Bott et al. Profil clinique, épidémiologique, thérapeutique et évolutif de la maladie de Vogt-Koyanagi-Harada: étude rétrospective d´une série de 10 cas au Maroc. Pan African Medical Journal. 2026;54:81. [doi: 10.11604/pamj.2026.54.81.53801]
Available online at: https://www.panafrican-med-journal.com//content/article/54/81/full
Case series 

Profil clinique, épidémiologique, thérapeutique et évolutif de la maladie de Vogt-Koyanagi-Harada: étude rétrospective d´une série de 10 cas au Maroc
Profil clinique, épidémiologique, thérapeutique et évolutif de la maladie de Vogt-Koyanagi-Harada: étude rétrospective d'une série de 10 cas au Maroc
Clinical, epidemiological, therapeutic, and outcomes profile of Vogt-Koyanagi-Harada disease: a retrospective study of 10 cases in Morocco
Zinab Bott1, ![]() Kadidiata Hamed Toure2, Amal El-Ouakhoumi1,
Kadidiata Hamed Toure2, Amal El-Ouakhoumi1, ![]() Jaouad Yousfi1,
Jaouad Yousfi1, ![]() Laila Benjilali1,
Laila Benjilali1, ![]() Mouna Zahlane1, Lamiaa Essaadouni1
Mouna Zahlane1, Lamiaa Essaadouni1
&Auteur correspondant
Nous avons mené une étude rétrospective, descriptive et analytique au CHU Mohamed VI de Marrakech (2017-2024) sur les patients retenus selon les critères standardization of uveitis nomenclature (SUN) 2021 et traité par corticothérapie et/ou immunosuppresseurs. Il s´agissait de décrire le profil épidémioclinique et l'influence des délais de prise en charge sur le pronostic visuel. Notre population était exclusivement féminine avec un âge moyen de 35,7 ans, le délai diagnostique médian était de 36 mois, délai d'introduction des immunosuppresseurs (IS) médian de 7,5 jours. On notait une amélioration significative de l'acuité visuelle (p=0,043). La précocité de l'immunomodulation après diagnostic est un facteur pronostique majeur pour compenser l'errance diagnostique.
We conducted a retrospective, descriptive, and analytical study at Mohammed VI University Hospital in Marrakech between 2017 and 2024, including patients diagnosed according to the 2021 standardization of uveitis nomenclature (SUN) criteria and treated with corticosteroids and/or immunosuppressive agents. The purpose was to describe the epidemioclinical profile and to assess the influence of delays in management on visual prognosis. The study population consisted exclusively of women, with a mean age of 35.7 years. The median time to diagnosis was 36 months, while the median interval between diagnosis and initiation of immunosuppressive therapy was 7.5 days. A significant improvement in visual acuity was observed (p=0.043). Early initiation of immunomodulatory therapy after diagnosis was identified as a major prognostic factor capable of mitigating the adverse effects of delayed diagnosis.
Key words: Vogt-Koyanagi-Harada syndrome, panuveitis, cyclophosphamide, immunosuppressive agents, visual acuity

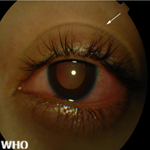
La maladie de Vogt-Koyanagi-Harada (VKH) est une panuvéite granulomateuse auto-immune rare et multisystémique, ciblant les tissus mélanocytaires oculaires, neurologiques et cutanés [1,2]. Sa physiopathologie repose sur une réponse lymphocytaire T CD4+ (Th1 et Th17) dirigée contre des auto-antigènes tels que la tyrosinase, survenant préférentiellement chez des individus génétiquement prédisposés portant l'allèle HLA-DRB1*0405 [3,4]. La gravité de cette affection réside dans son potentiel destructeur rapide: sans prise en charge thérapeutique immédiate, elle peut conduire à une cécité irréversible par atrophie chorio-rétinienne, décollements séreux rétiniens sévères et glaucome secondaire. Bien que le diagnostic ait été transformé par l'imagerie multimodale (OCT-EDI, angiographie au vert d'indocyanine) et les critères de classification du Standardization of Uveitis Nomenclature (SUN) 2021 [5,6], la prise en charge reste un défi. L'urgence thérapeutique est le pivot de la gestion clinique, car la fenêtre d'opportunité pour prévenir les séquelles est étroite [5,7].
Malgré ces avancées, les données sur les délais réels de prise en charge en contexte nord-africain restent limitées. Cette étude vise donc à décrire le profil épidémioclinique d'une cohorte marocaine, tout en évaluant l´efficacité du traitement sur l´acuité visuelle et à analyser l'influence des délais de prise en charge sur le pronostic visuel final.

Il s'agit d'une étude rétrospective, descriptive et analytique menée au sein du service de médecine interne du CHU Mohamed VI de Marrakech, au Maroc. L´étude a porté sur une période s'étendant de janvier 2017 à décembre 2024. L'étude a porté sur une cohorte monocentrique de 10 patientes (N=10) atteintes de la maladie de VKH, totalisant 19 yeux pour l'analyse de l'acuité visuelle. Les données ont été recueillies à partir des dossiers médicaux de patients ayant reçu un diagnostic de maladie de Vogt-Koyanagi-Harada selon les critères de classification SUN (2021). Les dossiers jugés incomplets ont été systématiquement exclus de l'analyse.
Les données collectées comprenaient: les variables démographiques (âge au diagnostic et sexe), les variables cliniques (classification clinique (forme incomplète, forme initiale) et présence d'atteintes extra-oculaires), les variables temporelles (délai de diagnostic, délai d´introduction des immunosuppresseurs (IS), durée de suivi et durée avant récidive) et les variables de pronostic visuel (acuité visuelle initiale (AVI) et finale (AVF)). L'acuité visuelle, initialement enregistrée selon l'échelle de Snellen, a été convertie en logarithme de l'angle de résolution minimum (LogMAR) pour permettre l'analyse statistique continue.
Pour les analyses statistiques, la normalité de la distribution des variables quantitatives a été évaluée par le test de Shapiro-Wilk. Les variables suivant une distribution normale sont exprimées en moyenne ± écart-type (s), tandis que celles à distribution non normale sont présentées en médiane (écart interquartile - IQR). L´efficacité du traitement a été analysée par le test t de student apparié pour comparer l´AV initiale et finale. Pour l'étude des facteurs pronostiques, le coefficient de corrélation de Spearman (ρ) a été utilisé pour mesurer la relation entre les délais (diagnostic et introduction des IS) et l'amélioration de l'AV (ΔAV). Enfin, le test de Mann-Whitney U a été employé pour comparer l´AV finale entre les groupes de diagnostic précoce (≤ 36 mois) et tardif (> 36 mois). Le seuil de signification statistique a été fixé à p < 0,05.
Caractéristiques de la population étudiée: la cohorte était exclusivement féminine (100%). L'âge moyen au diagnostic était de 35,7 ± 16,59 ans (extrêmes de 11 à 68 ans) avec une distribution normale confirmée par le test de Shapiro-Wilk (p=0,662) (Tableau 1).
Données cliniques et thérapeutiques: la maladie se présentait majoritairement sous une forme incomplète (9 cas, 90%). L'atteinte oculaire était bilatérale dans 90% des cas. Les manifestations cliniques les plus fréquentes étaient la panuvéite et le décollement rétinien séreux (DSR), observés chez 70% des patientes (Tableau 2). Sur le plan thérapeutique, toutes les patientes ont reçu une corticothérapie à forte dose associée à un agent immunosuppresseur, le cyclophosphamide étant l'agent le plus prescrit (60%). Le taux de récidive observé était de 40%, avec un délai moyen de survenue de 9,00 ± 5,66 mois après l'épisode initial.
Pronostic visuel et analyse statistique: l'acuité visuelle (AV) initiale moyenne était de 1,466 ± 0,999 LogMAR. À l'issue du suivi, une amélioration moyenne de l'AV de 0,511 LogMAR par œil a été constatée, une progression jugée hautement significative par le test t de student apparié. Un succès fonctionnel (AV finale ≤ 0,3 LogMAR) a été atteint pour 52,63% (11/19) des yeux (Tableau 3).
Analyse des facteurs pronostiques: l'analyse de corrélation de Spearman n'a révélé aucune corrélation significative entre le délai de diagnostic et l'amélioration de l'AV (ρ = -0,187, p = 0,415), ni entre le délai d'introduction des immunosuppresseurs et cette amélioration (p = 0,697). De même, la comparaison par le test de Mann-Whitney U de l'AV finale entre les groupes de diagnostic précoce (≤ 36 mois) et tardif (> 36 mois) n'a pas montré de différence statistiquement significative (p = 0,147). Néanmoins, l'AV médiane finale était meilleure dans le groupe précoce (0,26 LogMAR) par rapport au groupe tardif (0,52 LogMAR), suggérant une tendance clinique favorable.
Notre étude a révélé une cohorte exclusivement féminine (100%) de patientes atteintes de la maladie de VKH. Nous avons mis en évidence un paradoxe temporel notable: une errance diagnostique médiane prolongée de 36 mois, contrastant avec une prise en charge thérapeutique rapide après diagnostic, avec un délai médian d'introduction des immunosuppresseurs (IS) de 7,5 jours. Malgré une amélioration significative de l'acuité visuelle (p=0,043), nos résultats montrent un taux de récidive élevé de 40% et des succès fonctionnels (AV finale ≤ 0,3 LogMAR) atteignant 52,63%.
L´interprétation générale et la confrontation aux données existantes en ce qui concerne l'âge moyen au diagnostic (35,7 ans) concorde avec la littérature internationale [3]. L´exclusivité feminine (100%) de notre cohorte dépassent les ratios classiques de 2 femmes pour un homme, rapportés dans la littérature [3]. Cette spécificité suggère l'influence de facteurs hormonaux ou de susceptibilités génétiques propres à la population nord-africaine qui mériteraient des études génotypiques approfondies [8]. L'absence de corrélation significative entre le délai diagnostique et le pronostic visuel final (p=0,88) suggère que, même en cas d'errance prolongée, l'instauration rapide d'une immunomodulation permet de limiter les séquelles irréversibles. Ce résultat souligne que le temps écoulé après le diagnostic est un levier thérapeutique crucial, souvent plus déterminant que le retard diagnostique lui-même [9]. Par ailleurs, nos taux de succès fonctionnels inférieurs aux séries «initial-onset» confirment l'agressivité du phénotype dans les formes diagnostiquées tardivement. L'échec relatif des protocoles actuels et la prévention des récidives (40%) justifient une escalade thérapeutique précoce vers les anti-TNF alpha, conformément aux études démontrant leur efficacité supérieure dans les formes réfractaires [10].
Cette étude bien que menée dans un contexte monocentrique, fournit des données précieuses sur le profil épidémioclinique dans une population nord-africaine spécifique. Si la petite taille de l'échantillon impose une prudence quant à l'extrapolation, nos conclusions renforcent le consensus international sur l'urgence thérapeutique dans le VKH, tout en soulignant des particularités locales qui pourraient faire l'objet d'études multicentriques futures afin de valider nos observations à plus large échelle.
Limites: la principale limite de notre travail est la taille réduite de l'effectif (N=10), ce qui limite la puissance statistique de nos analyses et explique probablement la non-significativité de certains facteurs de gravité (p=0,07). De plus, le caractère rétrospectif de l'étude peut introduire des biais de sélection ou des données manquantes inhérentes à l'exploitation des dossiers médicaux.
Cette étude confirme l'efficacité des immunosuppresseurs dans la prise en charge de la maladie de Vogt-Koyanagi-Harada, comme en témoigne l'amélioration visuelle hautement significative observée dans notre cohorte. Le pronostic visuel fonctionnel obtenu, malgré une errance diagnostique médiane prolongée, souligne que la rapidité d'introduction de la thérapie immunosuppressive dès l'établissement du diagnostic est un levier thérapeutique crucial, potentiellement plus déterminant que la durée totale de la maladie avant la prise en charge. Enfin, la généralisation de l'imagerie multimodale (OCT-EDI) et l'application rigoureuse des critères de classification SUN 2021 demeurent essentielles pour réduire l'errance diagnostique et optimiser la précocité de la prise en charge dans notre contexte clinique.
Etat des connaissances sur le sujet
- Profil clinique: la maladie de Vogt-Koyanagi-Harada est reconnue comme une panuvéite granulomateuse auto-immune multisystémique sévère, nécessitant souvent une prise en charge immunosuppressive agressive;
- Défis diagnostiques: des retards diagnostiques prolongés sont fréquemment rapportés dans la littérature, menant souvent à des dommages structurels oculaires irréversibles si le traitement n'est pas initié rapidement;
- Thérapie standard: les corticoïdes systémiques restent le traitement de première intention, les agents immunomodulateurs (dont le cyclophosphamide) étant souvent réservés aux cas réfractaires ou aux présentations sévères.
Contribution de notre étude à la connaissance
- Validation clinique: cette étude apporte la preuve robuste qu'une initiation rapide de l'immunomodulation, même après un retard diagnostique significatif (médiane de 36 mois), peut améliorer de manière significative l'acuité visuelle ($p=0,043$);
- Données pronostiques: l'étude identifie l'intervalle entre le diagnostic et le début de l'immunomodulation (médiane de 7,5 jours) comme un facteur pronostique critique pour les résultats visuels;
- Prise en charge en vie réelle: en documentant l'utilisation efficace du cyclophosphamide dans une cohorte où 40% des patientes ont présenté des récidives, cette étude souligne la nécessité de stratégies d'escalade thérapeutique dans la gestion des formes sévères de VKH au sein de contextes cliniques spécifiques.
Les auteurs ne déclarent aucun conflit d'intérêts.
Zinab Bott: acquisition des données (cas 1), prise en charge clinique et revue de la littérature; Kadidiata Hamed Toure: prise en charge clinique (cas 2), acquisition des données, rédaction du manuscrit (version initiale); Amal El-Ouakhoumi: supervision de la prise en charge clinique; Jaouad Yousfi: supervision de la prise en charge clinique; Laila Benjilali: supervision senior, révision critique du manuscrit; Lamiaa Essaadouni: supervision senior, révision critique du manuscrit. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Les auteurs tiennent à remercier les patients pour leur coopération et pour avoir autorisé l'utilisation de leurs données cliniques dans le cadre de ce rapport de cas.
Tableau 1: paramètres épidémiologiques des 10 cas de Vogt-Koyanagi-Harada recrutés dans le service de médecine interne du CHU Mohamed VI Marrakech Maroc pour notre étude rétrospective s´étendant de janvier 2017 à décembre 2024
Tableau 2: paramètres cliniques des 10 cas de Vogt-Koyanagi-Harada recrutés dans le service de médecine interne du CHU Mohamed VI Marrakech Maroc pour notre étude rétrospective s´étendant de janvier 2017 à décembre 2024
Tableau 3: paramètres évolutifs des 10 cas de Vogt-Koyanagi-Harada (19 yeux) recrutés dans le service de médecine interne du CHU Mohamed VI Marrakech Maroc pour notre étude rétrospective s´étendant de janvier 2017 à décembre 2024
- Joye A, Suhler E. Vogt-Koyanagi-Harada disease. Curr Opin Ophthalmol. 2021 Nov 1;32(6):574-582. PubMed | Google Scholar
- Tayal A, Daigavane S, Gupta N. Vogt-Koyanagi-Harada Disease: A Narrative Review. Cureus. 2024 Apr 23;16(4):e58867. PubMed | Google Scholar
- Urzua CA, Herbort CP Jr, Takeuchi M, Schlaen A, Concha-Del-Rio LE, Usui Y et al. Vogt-Koyanagi-Harada disease: the step-by-step approach to a better understanding of clinicopathology, immunopathology, diagnosis, and management: a brief review. J Ophthalmic Inflamm Infect. 2022 May 12;12(1):17. PubMed | Google Scholar
- Albalawi AM, Al-Barry MA. Genetic variations in autoimmune genes and VKH disease. Int Ophthalmol. 2020 Nov;40(11):3175-3186. PubMed | Google Scholar
- Herbort CP Jr, Tugal-Tutkun I, Abu-El-Asrar A, Gupta A, Takeuchi M, Fardeau C et al. Precise, simplified diagnostic criteria and optimised management of initial-onset Vogt-Koyanagi-Harada disease: an updated review. Eye (Lond). 2022 Jan;36(1):29-43. PubMed | Google Scholar
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Vogt-Koyanagi-Harada Disease. Am J Ophthalmol. 2021 Aug:228:205-211. PubMed | Google Scholar
- O'Keefe GA, Rao NA. Vogt-Koyanagi-Harada disease. Surv Ophthalmol. 2017 Jan-Feb;62(1):1-25. PubMed | Google Scholar
- Bonnet C, Daudin JB, Monnet D, Brézin A. Maladie de Vogt-Koyanagi-Harada. J Fr Ophtalmol. 2017 Jun;40(6):512-519. PubMed | Google Scholar
- Rahman N, Artiaga JCM, Bouras K, Luis J, Rees A, Westcott M. Immunosuppressive therapy for Vogt-Koyanagi-Harada disease: a retrospective study and review of literature. J Ophthalmic Inflamm Infect. 2023 May 19;13(1):27. PubMed | Google Scholar
- Zhong Z, Wang J, Liao W, Xiang J, Cai T, Yang P. Effect of prednisone plus either adalimumab or cyclosporine on dermatological symptoms in Vogt-Koyanagi-Harada disease: Systemic outcomes from a randomized trial. J Am Acad Dermatol. 2025 Aug;93(2):415-422. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the JOURNAL_ABBREVIATION





Authors´ services
