
Le syndrome Sturge-Weber: à propos d´un cas
Meriem Doumiri, Mohamed Labied, Siham Salam, Dalal Laoudiyi, Kamilia Chbani, Lahcen Ouzidane
Corresponding author: Mohamed Labied, Service de la Radiologie Pédiatrique, Hôpital Harouchi, Casablanca, Morocco 
Received: 15 Jun 2020 - Accepted: 02 Jul 2020 - Published: 12 Aug 2020
Domain: Radiology,Pediatric neurology
Keywords: Sturge-Weber, angiome plan, imagerie par résonnance magnétique
©Meriem Doumiri et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Meriem Doumiri et al. Le syndrome Sturge-Weber: à propos d´un cas. Pan African Medical Journal. 2020;36:273. [doi: 10.11604/pamj.2020.36.273.24346]
Available online at: https://www.panafrican-med-journal.com//content/article/36/273/full
Case report 

Le syndrome Sturge-Weber: à propos d´un cas
Le syndrome Sturge-Weber: à propos d´un cas
Sturge-Weber syndrome: about a case
Meriem Doumiri1, Mohamed Labied1,&, Siham Salam1, Dalal Laoudiyi1, Kamilia Chbani1, Lahcen Ouzidane1
&Auteur correspondant
Le syndrome de Sturge-Weber (SWS) ou angiomatose encéphalo-faciale, est un syndrome neuro-cutané et oculaire congénital rare. Il comporte deux types de malformations: capillaire faciale congénitale à type d´angiome plan et capillaro-veineux lepto-méningé de localisation le plus souvent pariéto-occipitale homolatérale. La Neuroimagerie, essentiellement l´imagerie par résonnance magnétique (IRM), joue un rôle important dans l'établissement du diagnostic, idéalement avant l'apparition de complications neuro-oculaires. Nous rapportons le cas d´un enfant chez qui le SWS est suspecté devant la présence d´un angiome facial et d´une épilepsie pharmaco-résistante.
Sturge-Weber syndrome (SWS) or encephalofacial angiomatosis is a rare neurocutaneous and congenital ocular syndrome. It can cause two malformations: congenital facial capillary planar angioma and leptomeningal venous-capillary angioma (most often parieto-occipital homolateral angioma). Neuroimaging, in particular magnetic resonance imaging (MRI), plays an important role in the diagnosis, ideally before the occurrence of neuro-ocular complications. We report the case of a child in whom SWS was suspected based on facial angioma and pharmaco-resistant epilepsy.
Key words: Sturge-Weber, planar angioma, MRI

Le syndrome de Sturge-Weber (SWS) ou angiomatose encéphalo-faciale, est un syndrome neuro-cutané et oculaire congénital rare. Il comporte deux types de malformations: capillaire faciale congénitale à type d´angiome plan et capillaro-veineux lepto-méningé de localisation le plus souvent pariéto-occipitale homolatérale. La tomodensitométrie et l´imagerie par résonnance magnétique sont considérées comme la clé diagnostic de ce syndrome.

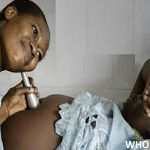
Nous rapportons le cas d´un enfant âgé de 6 ans, de sexe masculin, admis au service de radiologie pour le bilan d´une épilepsie pharmaco-résistante. On n'a pas noté d'épilepsie dans les antécédents familiaux, pas de notion de consanguinité, et l'enfant avait jusqu'à ce jour un bon développement psychomoteur. A l´examen, le patient était conscient, apyrétique, sans déficit sensitivo moteur. A l´examen clinique, on a objectivé un angiome plan cutané au niveau fronto-orbitaire gauche (Figure 1). Une IRM fut réalisée (Figure 2) mettant en évidence un épaississement du plexus choroïde gauche ainsi qu'une prise de contraste accentuée et gyriforme des sillons corticaux pariéto-occipitaux qui sont discrètement élargies témoignant de cette atrophie cérébrale homolatérale. Des dépôts calciques ont été détectées sur la séquence T2*. Ainsi le diagnostic d´un syndrome de Sturge Weber fut évoqué, et le patient a été adresser en consultation ophtalmologique.
Le syndrome de Sturge-Weber (SWS) est une phacomatose neuro-cutanée et oculaire, rare et sporadique, qui résulte d´une malformation du système vasculaire fœtal entraînant une anoxie corticale [1]. Dans sa forme complète ce syndrome comporte: une malformation capillaire faciale congénitale à type d´angiome plan, un angiome capillaro-veineux lepto-méningé de localisation le plus souvent pariéto-occipitale, avec atrophie cérébrale et calcifications sous-corticales, et des anomalies oculaires (glaucome et maladie vasculaire choroïdienne) [2,3]. L'anomalie physiopathologique fondamentale à la base de l'angiomatose leptoméningée est l'échec du développement normal des veines au niveau de la corticale et la persistance résultante du complexe vasculaire primitif fœtal [4]. Pendant de nombreuses années, ce syndrome était défini par une distribution selon le territoire du nerf trijumeau. Récemment, en 2014, Waelchli et al. ont proposé que la distribution de ce syndrome suit la distribution des vaisseaux sanguins embryonnaires du visage plutôt que celle du nerf trijumeau [5]. La présence de ces vaisseaux anormaux conduit à une altération de la perfusion cérébrale et à une ischémie progressive du parenchyme cérébral sous-jacent. Cela sera encore aggravé dans les cas de crises convulsives non contrôlées.
Dans 80% des cas, il existe une atteinte hémisphérique cérébrale unilatérale, ce qui explique la fréquence élevée d'hémiatrophie cérébrale chez les patients atteints du SWS [6]. Les manifestations cliniques du syndrome comprennent une épilepsie réfractaire au traitement, voire une hémiplégie, une hémianopsie et un retard mental. La neuroimagerie joue un rôle crucial dans l'établissement du diagnostic, idéalement avant l'apparition de complications neuro-ophtalmologiques. L´IRM aide à établir le diagnostic et à évaluer l´atteinte intracrânienne, mais il n´existe pas de consensus quant aux personnes à dépister, au moment optimal de l´imagerie, à la sensibilité de l´IRM ou à l´avantage global de l´identification des anomalies vasculaires à risque. La présence d'angiomatose leptoméningée a été considérée comme une caractéristique directe (impliquant généralement le cortex pariétal ou occipital postérieur ipsilatéral aux signes faciaux) [7]. L'IRM est la modalité d'imagerie idéale pour rechercher cette entité. Tout l'étendue de l'angiome pial est visualisé sous forme de prise de contraste dans les espaces sous-arachnoïdiens et des gyrus en regard.
L'atrophie des zones concernées caractérisent les cas avancés. La séquence pondérée en écho de gradient (T2*) est utile pour visualiser les dépôts calciques cortico-sous-corticaux. On peut retrouver un élargissement des veines profondes dû à une dysgénésie du système veineux superficiel, entraînant une dérivation du sang. Un épaississement du plexus choroïde peut également être retrouvé, attribué à cette dérivation dans les veines profondes [8]. Au cours des premières années de la vie, sur la séquence de perfusion, cette malformation vasculaire tend à devenir hypo perfusée, par altération du drainage veineux [9]. La tomodensitométrie montrent des calcifications dans la substance blanche sous-corticale et dans le cortex adjacent gyriforme (tram tracksign). La cause de ces calcifications est attribuée à une ischémie chronique due à un drainage veineux altéré. Un large spectre de malformations des développements corticaux est observé chez les patients atteints de SWS, allant de la polymicrogyrie et de la schizencéphalie à la dysplasie corticale focale [10].
Le diagnostic du syndrome de Sturge-Weber est suspecté chez les patients présentant un angiome du front. Face à cette suspicion, un examen ophtalmologique et une IRM avec injection de gadolinium devraient être réalisés pour permettre un diagnostic précoce et réduire les complications ophtalmologiques et cérébrales.
Les auteurs ne déclarent aucun conflit d´intérêts.
Tous les auteurs ont contribué à la conduite de ce travail de recherche. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: angiome plan fronto-orbitaire gauche
Figure 2: (A) séquence axiale T2: atrophie pariéto-occipitale gauche associée à une dédifférenciation substance blanche-substance grise; (B) séquence axiale T2(astérique): vides de signaux linéaires cortico-sous-corticaux en rapport avec des dépôts calciques; (C) séquence T1 écho de spin après injection de contraste: aspect épaissi du plexus choroïde gauche, et prise de contraste lépto-méningée en rapport avec un angiome pial
- Rumboldt Z, Castillo M, Huang B. Brain imaging with MRI and CT: an image pattern approach. Cambridge Medicine. 2012:428. Google Scholar
- Huckman MS. Imaging of cerebral phakomatoses: nicer course neuroradiol. Pediatr Radiol. 1995;25:28-29.
- Enjolras O, Riché MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985;76(1):48-51. PubMed | Google Scholar
- Griffiths PD. Sturge-Weber syndrome revisited: the role of neuroradiology. Neuropediatrics. 1996;27(6):284-294. PubMed | Google Scholar
- Waelchli R, Aylett SE, Robinson K, Chong WK, Martinez AE, Kinsler VA. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014;171(4):861-867. PubMed | Google Scholar
- Terdjman P, Aicardi J, Sainte-Rose C, Brunelle F. Neuroradiological findings in Sturge-Weber syndrome (SWS) and isolated pial angiomatosis. Neuropediatrics. 1991;22(3):115-120. PubMed | Google Scholar
- Benedikt RA, Brown DC, Walker R, Ghaed VN, Mitchell M, Geyer VA. Sturge-Weber syndrome: cranial MR imaging with Gd- DTPA. AJNR Am J Neuroradiol. 1993;14(2):409-415. PubMed | Google Scholar
- Zhou J, Li NY, Zhou XJ, Wang JD, Ma HH, Zhang RS. Sturge-Weber syndrome: a case report and review of literatures. Chin Med J (Engl). 2010 Jan 5;123(1):117-121. PubMed | Google Scholar
- Lin DDM, Barker PB, Hatfield LA, Comi AM. Dynamic MR perfusion and proton MR spectroscopic imaging in Sturge- Weber syndrome: correlation with neurological symptoms. J Magn Reson Imaging. 2006;24(2):274-281. PubMed | Google Scholar
- Uduma FU, Emejulu JK, Motah M, Okere PC, Ongolo PC, Muna W. Differential diagnoses of cerebral hemiatrophy in childhood: a review of literature with illustrative report of two cases. Glob J Health Sci. 2013 May;5(3):195-207. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
PlumX Metrics
Le syndrome Sturge-Weber: à propos d´un casRecently from the PAMJ





Authors´ services
