
Management of jugular tympanic paraganglioma: a case report
Mehdi Ferjaoui, Naourez Kolsi, Wiem Boughzala, Ons Kharrat, Rachida Bouatay Khaled Harrathi, Amel Elkorbi, Jamel Koubaa
Corresponding author: Mehdi Ferjaoui, Ear, Nose, Thorax Department of Fattouma Bourguiba, University Hospital of Monastir, Resaerch Laboratory LR18Sp08, Monastir, Tunisia 
Received: 23 Apr 2021 - Accepted: 04 Nov 2022 - Published: 02 Dec 2022
Domain: Otolaryngology (ENT)
Keywords: Jugular tympanic paraganglioma, tinnitus, medical imaging, surgery, case report
©Mehdi Ferjaoui et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mehdi Ferjaoui et al. Management of jugular tympanic paraganglioma: a case report. Pan African Medical Journal. 2022;43:166. [doi: 10.11604/pamj.2022.43.166.29457]
Available online at: https://www.panafrican-med-journal.com//content/article/43/166/full
Case report 

Management of jugular tympanic paraganglioma: a case report
Management of jugular tympanic paraganglioma: a case report
Mehdi Ferjaoui1,&, Naourez Kolsi1, ![]() Wiem Boughzala1, Ons Kharrat1, Rachida Bouatay1, Khaled Harrathi1, Amel Elkorbi1, Jamel Koubaa1
Wiem Boughzala1, Ons Kharrat1, Rachida Bouatay1, Khaled Harrathi1, Amel Elkorbi1, Jamel Koubaa1
&Corresponding author
Paragangliomas could be localized from the skull base to the pelvic floor. Tympanic localization represents the most common benign tumor of the middle ear. Diagnosis is based on clinical signs with a great contribution of radiology. A 40-year-old male presented with isolated tinnitus of the right ear evolving for 18 months. Examination revealed a red bulging right-sided tympanic membrane and a conductive hearing loss. Tomodensitometry and Magnetic resonance imagery showed findings in favor of a right jugular tympanic paraganglioma. The tumor was classified type B according to FISCH classification. The patient underwent surgery consisting in tympanotomy using a retro auricular access route. The postoperative course was uneventful. There was no recurrence during the one-year follow-up. Jugular tympanic paraganglioma diagnosis is guided by a combination of epidemiological, clinical and radiological features. Treatment is still not consensual, but surgery still have its indications in localized forms of head and neck paragangliomas (HNP´s).

Paragangliomas could be localized from the skull base to the pelvic floor. They are embryologically derived from the neural crest and arise from paraganglia cells within a highly vascular environment. They are classified by their location. Head and neck paragangliomas (HNPs) originate from the parasympathetic tissue, with the carotid body, vagal body and jugular tympanic region are the most common locations [1]. Tympanic paraganglioma represents the most common benign tumor of the middle ear. Diagnosis is based on clinical signs with a great contribution of imagery technics. Treatment is not consensual, but surgery still has its place in some forms of HNPs.

We report the case of a jugular tympanic paraganglioma, diagnosed and treated in our ENT department in 2019.
Patient information: a fourty-year-old male, smoker, presented in consultation with gradually increasing, unilateral, right tinnitus, evolving for 18 months, associated with hear loss. Tinnitus was swishing as reported by the patient, synchronous with pulse, and responsible of an important discomfort, causing concentration difficulties and insomnia. The patient did not report any otorrhea, otalgia or vertigo. He did not report any sign for the left ear. His past medical history was normal for any cerebrovascular accident, migraine, headaches, head and neck trauma, or radiation exposure. He had no family history of paragangliomas nor multiple endocrine neoplasia. He did not any past interventions.
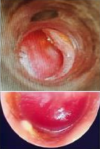
Clinical findings: otoscopic examination, using a rigid endoscope 0° (Figure 1), revealed a red pulsatile swelling behind the lower part of the right ear-drum. The Weber test was lateralized to the pathologic ear, and the Rinne test was negative. Carotid manual pressure temporarily stopped the tinnitus. The rest of the physical examination showed no neurological deficits, particularly no defects corresponding to one or more of IXth, Xth, XIth and XIIth cranial nerves.
Timeline of current episode : the symptoms had been evolving for 18 months with a gradual exacerbation.
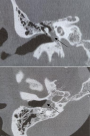
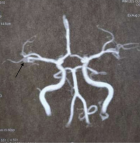
Diagnostic assessment: pure-tone audiometry showed a right conductive hearing loss of 30dB. No abnormalities were detected on the left side. Temporal bones Computerized tomography (CT), axial and coronal sections, without contrast, showed a nonspecific, well-defined, 7 mm right nodular mass of soft tissue, arising from the hypotympanum and overlying the cochlear promontory (Figure 2). It reaches the oval and the round foramina without any bone erosion. The ossicular chain, the facial nerve canal and the inner ear were intact. MRI showed a 15 mm mass of the right tympanic cavity, with a “salt and pepper” pattern of hypo intensity and hyper intensity on T1-weighted images, relatively higher in intensity on T2-weighted images, and strongly enhanced after gadolinium injection (Figure 3). On diffusion-weighted images, it appeared in hypersignal. The lesion reaches the tympanic membrane and expands through the aditus ad antrum into the mastoid antrum. These findings appeared to be consistent with jugular tympanic paraganglioma. There were no signs of invasion of the skull base. MRI was completed with angio-MRI sequences showing a hyper vascular lesion corresponding to the glomus tympanicum tumor (Figure 4).
Diagnosis: considering the radiological examination, the paraganglioma was classified type B according to FISH classification of jugular tympanic paragangliomas (Table 1).
Prognostic characteristics: the Type B paraganglioma is still localized to middle ear, surgery could be complete and successful for our case.
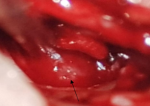
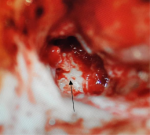
Therapeutic interventions: after considering the size of the tumor and its type, the decision was to proceed to a surgical treatment without primary embolization. The patient underwent a tympanotomy using a retro auricular access route. On per-operative examination, the tumor was hyper vascular, easily bleeding, emerging from the hypotympanum, filling the tympanic cavity and occluding the Eustachian tube (Figure 5). The intervention lasted two hours. The lesion was totally excised (Figure 6, Figure 7) without mastoidectomy which was judged unnecessary.
Follow-up and outcome of interventions: the postoperative course was uneventful. Anatomopathological microscopic examination revealed a benign tumor with cells arranged in nests and alveolar patterns separated by prominent blood vessels. Cells were with monomorphic fine granular chromatism nuclei and eosinophilic granular cytoplasm. Immunostaining for synaptophysin was positive and PS100 marked lenticular sus cells. The histopathologic findings thus confirmed diagnosis of paraganglioma. There was no recurrence during the one-year follow up.
Patient perspective: the patient should share their perspective in one to two paragraphs on the treatment(s) they received.
Informed consent: the patient was informed by the different modalities of treatments and was consenting for surgery.
Head and neck paragangliomas (HNPs) represent neuroendocrine tumors developed from chromaffin cells within paraganglionic tissues of the autonomic nervous system [1]. They represent 0.03% of all human tumors and less than 0.5% of head-and-neck tumors. Their annual incidence is 0.001% [2,3]. Paraganglioma localizations vary widely. In Erickson´s series of 204 HNPs, 57% were localized in the carotid body, 30% were tympano-jugular and 13% vagal [4]. The gender ratio (F:M) is 2:1. The average age of manifestation is the 5th decade [1]. Paragangliomas could occur sporadically or as hereditary tumors, especially in multiple endocrine neoplasia type II [1,5]. These forms appear in younger ages and present more multicentricity and bilateralism [1]. Pulsatile tinnitus is the most suggestive symptom of JTP. Progressive unilateral hearing loss could be caused by an ossicular lysis or a cochlear invasion. These signs were absent in our case. Less frequent manifestations are otalgia, vertigo, and neurological deficits (hoarseness, dysphagia and facial nerve palsy) [6]. Considering the slow growth rate of these tumors (0.8mm/year), an important consultation delay was reported [7]. Otoscopy reveals a retro-tympanic reddish-blue pulsatile mass [8], producing the “Rising Sun” appearance. Neurological examination may find Vernet syndrome secondary to invasion of the jugular foramen, with paralysis of the IXth, Xth and XIth cranial nerves. Associated XIIth palsy forms Collet-Sicard syndrome [9]. On audiometry, a conductive hearing loss was found in all JTP, just like in our case. A sensorineural hearing loss could be associated in large tumors with intracranial extension [10]. Screening for catecholamines and their metabolites in urine and plasma in all cases of glomus tympanic tumor was previously reported. However, since catecholamine secretion occurs in only 1% to 4% of cases, recent studies recommend these investigations only in hereditary and multifocal forms, or in case of suggestive symptoms: diarrhea, flushing, hypertension and palpitations [11,12]. This explains why they were omitted in our case.
Imagery is necessary to diagnose HNP, classify the tumor and establish a treatment plan. Temporal bone CT and MRI are complementary [11]. Small JTP are seen on CT as well-defined intratympanic masses without significant bone involvement. Larger tumors can surround the ossicles, extend to the Eustachian tube, through the aditus to the mastoid and through the tympanic membrane to the external auditory canal. They could reach skull base neural foramina. CT scan has higher sensitivity to differentiate between tympanic glomus and jugular glomus by analyzing the bone plate separating the tympanic cavity from the jugular bulb. It could detect multicentric tumors along the vagus nerve and the carotid bifurcation [11]. The absence of bone destruction represents the most important differential feature with cholesteatoma. MRI is excellent to evaluate the soft tissue component of the tumor and its intracranial extension. The “salt-and-pepper” pattern of high and low signals on T2-weighted sequences and the contrast enhancement on T1-weighted sequences are nearly pathognomonic of paragangliomas [5,12]. In our case, brain MRI confirmed the diagnosis and classified it Type 2 of FISCH classification. Largely abandoned angiography could be indicated before embolization or if diagnosis is uncertain [11,12]. The 18F-dihydroxyphenylalanin positron emission tomography represents a promising diagnostic tool for smaller than 1cm paraganglioma´s [5]. Radiological examination allows tumor staging according to “FISCH classification” to establish adapted treatment modalities [11]. They include abstention, surgical resection, embolization and radiotherapy. Surgery remains the only radical treatment for JTPs. The surgical approach depends on the JTP´s localization and extension, it could be trans-canal or postauricular [7,12]. Preoperative embolization decreases bleeding during surgery. Post-operative complications are hearing loss, facial weakness, tympanic membrane perforation (1.7%), acquired cholesteatoma (1.7%), surgical-site infection (1.7%) and cerebrospinal fluid leak (0.9%) [12]. Jackson reports mortality rates of 0% to 4% [13].
Radiotherapy, used exclusively or postoperatively, could assure long-term tumor control. The recommended dose is 45-50 Gy fractioned over 5 weeks [7]. A study by Gilbo reported a local control of 96%, a cause-specific survival of 97% and an overall survival of 72% after a 10-year follow-up. No patient experienced severe complications [14]. Stereotactic radiotherapy treatment is an interesting alternative. It is administered in one fraction and applies minimal radiation to the adjacent normal tissues [7]. Close observation was described in literature as a possible strategy, with clinical and radiological evaluation every 6 to 12 months, extendable in case of stability [5,7,12,15]. It is considered in case of significant comorbidities, for elderly, or for pauci-symptomatic patients. A multidisciplinary consultation is useful to choose the most adapted treatment protocol. The patient´s clarified-consent is compulsory [7]. For Fisch class A and B, like our case, exclusive surgical resection is the gold standard. Fisch class C and D require postoperative radiotherapy. Embolization 24 to 48 hours before surgery is indicated for Fisch class B, C and D. As a palliative setting, it could be used to slow down tumor´s growth [5,7]. Ivan and Springate [16,17] describe tumor control rates of 86% after exclusive surgery, 69% after subtotal tumor resection, 71% to 90% after subtotal resection combined with adjuvant RT and 93% to 95% after exclusive RT. These findings go in pair with Miller´s conclusions [18,19]. In combined procedures, no recurrences were reported after a mean follow-up of 30.4 months [12].
Jugular tympanic paraganglioma are challenging rare lesions. Precise clinical assessment, preoperative imaging and interdisciplinary discussions are the basis of an adequate management of HNPs. Function-preserving therapy seems to be the most adapted approach, even if, to date, there are still no consensual recommendations in literature.
The authors declare no competing interests.
All the authors read and approved the final version of the manuscript.
Table 1: the FISCH classification of jugulotympanic paragangliomas
Figure 1: the bulging of the tympanic membrane produced by a glomus tumor: A) otoscopic examination; B) endoscopic examination showing a red pulsatile swelling behind the lower part of the right ear-drum
Figure 2: temporal bones computerized tomography (CT), coronal (A) and axial (B) sections, without contrast, showing a nonspecific, well-defined, 7 mm right nodular mass of soft tissue, arising from the hypotympanum and overlying the cochlear promontory
Figure 3: MRI showing a 15 mm mass of the right tympanic cavity, with a "salt and pepper" pattern of hypo intensity and hyper intensity on T1-weighted images (A), relatively higher in intensity on T2-weighted images (B), and strongly enhanced after gadolinium injection (C)
Figure 4: angio-MRI sequences showing a hyper vascular lesion
Figure 5: per-operative view: the tumor was hyper vascular, easily bleeding, emerging from the hypotympanum, filling the tympanic cavity and occluding the Eustachian tube
Figure 6: postoperative cavity after complete excision of the tumor
Figure 7: specimen: a un cm red mass
- Valero C, Ganly I, Shah JP. Head and neck paragangliomas: 30-year experience. Head Neck. 2020;42(9):2486-2495. PubMed | Google Scholar
- Singh S, Madan R, Singh MK, Thakar A, Sharma SC. Head-and-neck paragangliomas: an overview of 54 cases operated at a tertiary care center. South Asian J Cancer. 2019;08(04):237-240. PubMed | Google Scholar
- Wasserman PG, Savargaonkar P. Paragangliomas: classification, pathology, and differential diagnosis. Otolaryngol Clin North Am. 2001;Otolaryngol Clin North Am. 2001 Oct;34(5):845-62, v-vi. PubMed | Google Scholar
- Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86(11):5210-5216. PubMed | Google Scholar
- Boedeker CC, Ridder GJ, Schipper J. Paragangliomas of the head and neck: diagnosis and treatment. Fam Cancer. 2005;4(1):55-59. PubMed | Google Scholar
- Harati A, Schultheiß R, Rohde S, Deitmer T. Disease and treatment-related sequelae in patients with complex jugulotympanic paraganglioma. J Clin Med. 2018 Mar 10;7(3):51. PubMed | Google Scholar
- Moore MG, Netterville JL, Mendenhall WM, Isaacson B, Nussenbaum B. Head and neck paragangliomas: an update on evaluation and management. Otolaryngol-Head Neck Surg Off J Am Acad Otolaryngol-Head Neck Surg. 2016;154(4):597-605. PubMed | Google Scholar
- Jehangir A, Pathak R, Shaikh B, Salman A, Fareedy SB, Qureshi A et al. Jugulotympanic paraganglioma: a rare cause of vertigo. Am J Case Rep. 2015;16:228-231. PubMed | Google Scholar
- Khalid S, Zaheer S, Khalid M, Zaheer S, Raghuwanshi RK. Collet-Sicard syndrome secondary to a large glomus jugulotympanicum. Ann Saudi Med. 2013;33(4):407-410. PubMed | Google Scholar
- Baguley DM, Irving RM, Hardy DG, Harada T, Moffat DA. Audiological findings in glomus tumours. Br J Audiol. 1994;28(6):291-297. PubMed | Google Scholar
- Noujaim SE, Pattekar MA, Cacciarelli A, Sanders WP, Wang AM. Paraganglioma of the temporal bone: role of magnetic resonance imaging versus computed tomography. Top Magn Reson Imaging TMRI. 2000;11(2):108-122. PubMed | Google Scholar
- Sweeney AD, Carlson ML, Wanna GB, Bennett ML. Glomus tympanicum tumors. Otolaryngol Clin North Am. 2015;48(2):293-304. PubMed | Google Scholar
- Jackson CG, McGrew BM, Forest JA, Netterville JL, Hampf CF, Glasscock MEI. Lateral skull base surgery for glomus tumors: long-term control. Otol Neurotol. 2001;22(3):377-382. PubMed | Google Scholar
- Gilbo P, Morris CG, Amdur RJ, Werning JW, Dziegielewski PT, Kirwan J et al. Radiotherapy for benign head and neck paragangliomas: a 45-year experience. Cancer. 2014;120(23):3738-3743. PubMed | Google Scholar
- Künzel J,Iro H, Hornung J, Koch M, Bras C, Klautke G et al. Function-preserving therapy for jugulotympanic paragangliomas: a retrospective analysis from 2000 to 2010. The Laryngoscope. 2012;122(7):1545-1551. PubMed | Google Scholar
- Ivan ME, Sughrue ME, Clark AJ, Kane AJ, Aranda D, Barani IJ et al. A meta-analysis of tumor control rates and treatment-related morbidity for patients with glomus jugulare tumors. J Neurosurg. 2011;114(5):1299-1305. PubMed | Google Scholar
- Springate S, Haraf D, Weichselbaum R. Temporal bone chemodectomas:comparing surgery and radiation therapy. Oncology (Williston Park). 1991 Apr;5(4):131-7,discussion 140, 143. PubMed | Google Scholar
- Miller JP, Semaan M, Einstein D, Megerian CA, Maciunas RJ. Staged gamma knife radiosurgery after tailored surgical resection: a novel treatment paradigm for glomus jugulare tumors. Stereotact Funct Neurosurg. 2009;87(1):31-36. PubMed | Google Scholar
- Miller JP, Semaan MT, Maciunas RJ, Einstein DB, Megerian CA. Radiosurgery for glomus jugulare tumors. Otolaryngol Clin North Am. 2009;42(4):689-706. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures



Article metrics
Recently from the PAMJ








Authors´ services
