
Coronavirus disease 2019 (COVID-19) pathogenesis: a concise narrative review
Hamza Elhamzaoui, Houssam Rebahi, Abdelhamid Hachimi
Corresponding author: Hamza Elhamzaoui, Critical Care Department, Mohammed VIth University Hospital of Marrakech, Cadi Ayyad University, Marrakech, Morocco 
Received: 15 May 2020 - Accepted: 01 Aug 2020 - Published: 03 May 2021
Domain: Infectious disease,Intensive care medicine
Keywords: Coronavirus, SARS-CoV-2, COVID-19, pathogenesis
©Hamza Elhamzaoui et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hamza Elhamzaoui et al. Coronavirus disease 2019 (COVID-19) pathogenesis: a concise narrative review. Pan African Medical Journal. 2021;39:8. [doi: 10.11604/pamj.2021.39.8.23546]
Available online at: https://www.panafrican-med-journal.com//content/article/39/8/full
Review 

Coronavirus disease 2019 (COVID-19) pathogenesis: a concise narrative review
Coronavirus disease 2019 (COVID-19) pathogenesis: a concise narrative review
Hamza Elhamzaoui1,&, ![]() Houssam Rebahi1,
Houssam Rebahi1, ![]() Abdelhamid Hachimi1
Abdelhamid Hachimi1
&Corresponding author
SARS-CoV-2 is the third zoonotic coronavirus. Since December 2019, it has spread through the globe and infects more than four million patients (as of May 10th, 2020). The disease was named coronavirus disease 2019 (COVID-19) by the World Health Organization (WHO). It involves many organs and systems in the human organism. We aimed to describe the pathogenesis of the COVID-19.

Of the coronaviridae family, human coronaviruses (HCoVs) include six strains. The first HCoV, named B814, was identified in 1965 in nasal swabs in patients with cold [1]. Since then, more HCoVs have been isolated: HCoV-229E (229E), HCoV-OC43 (OC43), HCoV-NL63 (NL63), HCoV-HKU1 (HKU1), severe acute respiratory syndrome coronavirus (SARS-CoV-2) and Middle East respiratory syndrome coronavirus (MERS-CoV) [2]. They cause primarily upper respiratory and gastrointestinal infections, which vary from mild (common cold) to severe acute respiratory syndrome with possible multi-organ failure [3]. SARS-CoV discovered during the SARS epidemic, started from Guangdong province in Southern China in November 2002 and spread to other countries in Asia, North America and Europe [4,5]. While, since 2012, MERS-CoV is responsible for a progressing outbreak in the Middle East [6]. After SARS-CoV and MERS-CoV, the third zoonotic coronavirus, novel coronavirus (2019-nCoV) has spread and infected more than four million persons worldwide (as of May 10th, 2020), since December 2019, from Wuhan, China [7]. The International Committee on Taxonomy of Viruses named 2019-nCoV as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The World Health Organization titled the disease caused by SARS-CoV-2 as coronavirus disease 2019 (COVID-19) [8-10]. Chan et al. Hui et al. Zhou et al. and Wu et al. suggested that the origin of SARS-CoV-2 was bats but the intermediate host remains unknown [11-14].
Since the first case, COVID-19 could be qualified as a “great imitator” because it is not a simple lung disease. It can cause more severe impairment such as heart rhythm problems, heart failure, acute kidney failure, hemorrhagic stroke, seizures, meningitis Guillain-Barre syndrome, or thromboembolic events [15,16]. When the virus gains the body via eyes, nose, or mouth, it invades cells by connecting to ACE2 receptors, which are found in the organs. In this review, we aimed to set out SARS-CoV-2 pathogenesis.

We performed a literature search in multiple databases dedicated to the literature about COVID-19: NEJM database [17], ESICM database [18], Elsevier information center [19], and Medscape center [20]. The keywords used in the research were coronavirus, SARS, MERS, SARS-CoV-2, MERS-CoV, SARS-CoV-1, SARS-CoV-2, 2019-nCoV and COVID-19.
Genetic predispositions: several factors make some people susceptible to the coronavirus and present phenotypes ranging from asymptomatic to severe forms of COVID-19 with acute respiratory distress syndrome with possible multi-organ failure. Gralinski et al. suggested that the alteration of the function of the innate-immune modulatory gene Ticam2 makes rodents highly susceptible to the coronavirus. This gene, Ticam2, codes for a helper protein in the activation of a family of receptors (TLR, for toll-like receptor) involved in the mechanisms of innate immunity [21]. Zhao et al. highlighted the second component [22]. They showed that blood group A is linked to a higher risk of acquiring COVID-19 (OR 1.279; 95% CI 1.136-1.440; p<0.001) and death (OR 1.482; 95%CI 1.113-1.972; p=0.008,) compared with other blood groups. Cheng et al. expressed the same findings for SARS-CoV-2 in 2005 [23]. The third genetic component points out the relationship between human leukocyte antigen (HLA) and the risk to contract and the severity of COVID-19. Indeed, the susceptibility to SARS-CoV-2 is associated with HLA-B*46:01 [24], as previously expressed by Lin et al. for SARS-CoV-2 [25].
The fourth component is related to the angiotensin-converting enzyme 2 (ACE2). According to Li et al. ACE2 is the functional receptor of SARS-CoV-2 [26]. Effectively, Hamming et al. reported that this receptor is plenteous in the human epithelia of the lung and small intestine [27], with its presence in the vascular endothelium. Experimentally, as outlined by Kuba et al. [28], the infected ACE2 knockout mice are resistant to virus infection and the virus titers are 105 fold lower than those isolated from the lung of SARS-CoV-2 infected wild-type mice, without evidence of inflammation in the lung histology. Recently, Yan et al. [29], Zhang et al. [30] and Wan et al. [31] stated that ACE2 is involved in the pathogenesis of SARS-CoV-2. To find out more about the genes that influence SARS-CoV-2 infection, some projects are underway [32,33].
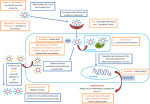
Pulmonary involvement: angiotensin-converting enzyme 2 (ACE2) is the host receptor through which SARS-CoV-2 enters cells (Figure 1) [34]. In normal lung tissue, ACE2 is mainly expressed by type I and type II alveolar epithelial cells. It was reported that 83% of type II alveolar cells expressed ACE2. Therefore, COVID-19 infection causes damages to most type II alveolar cells. After alveolar cell injury, transforming growth factor-β (TGF-β) is released in the tissue to promote lung repair. Virus infection often leads to excessive activation of the TGF-β pathway, which leads to the occurrence of pulmonary fibrosis [35,36]. In the early phases of the lung pathology of COVID-19, Tian et al. described findings of edema, proteinaceous exudate, focal reactive hyperplasia of pneumocytes with patchy inflammatory cellular infiltration and multinucleated giant cells in a pathologic examination of infected lungs [37]. Even though the COVID-19 meets the acute respiratory distress syndrome (ARDS) Berlin definition [38], it is a disease with distinctive phenotypes and can even be part of the ARDS mimics described by Aublanc et al. [39]. Its main characteristic is the dissociation between the severity of the hypoxemia and the maintenance of relatively good respiratory mechanics [40]. To conceptualize this phenomenon, Gattinoni L et al. [41] hypothesized a sequence of events: SARS-CoV-2 infection causes a modest local subpleural interstitial edema located between lung structures with different elastic properties [42]. The normal response is to increase minute ventilation, by increasing the tidal volume [43], which is associated with a more negative intrathoracic inspiratory pressure. However, the near-normal compliance explains why some of the patients present without dyspnea as the patient inhales the volume he expects.
Two peculiar phenotypes can be distinguished: type L, characterized by Low elastance (i.e. high compliance), low ventilation to perfusion ratio, low lung weight and low recruitability. These patients may remain unchanging for a period and then improve or worsen. The possible feature that can determine the evolution of the disease, other than severity, is the depth of the negative intrathoracic pressure associated with the increased tidal volume in spontaneous breathing. The increased lung permeability due to inflammation, associated with this negative intrathoracic pressure, causes interstitial lung edema. As the edema increases, the lung weight increases, until eventually the gas volume in the lung decreases and the tidal volumes decrease. This phenomenon leads to a transition from type L, to type H, which is characterized by high elastance, high right-to-left shunt, high lung weight and high recruitability. These patients likely develop self-inflicted ventilator-induced lung injury [40,41]. These different COVID-19 patterns depend on the interaction between three factors [41]: 1) the severity of the infection, the host response, and comorbidities; 2) the ventilatory response to hypoxemia and 3) the time between the beginning of the disease and the hospitalization.
Hematological disorders: besides, in the early phases of infection, ACE-2 consumption by viral entry is predicted to increase local angiotensin II concentration. Among the known effects of angiotensin II are vasoconstriction, endothelial activation, and pro-inflammatory cytokine release [44]. Consequently, viral injury, disordered cytokine release and platelet activation by angiotensin II induce localized microvascular inflammation, which triggers endothelial activation, leading to vasodilation and pro-thrombotic conditions. The procoagulant state has also long been recognized as part of ARDS pathophysiology, demonstrated by the identification of diffuse pulmonary endothelial injury associated with platelets' activation, macro- and micro-thrombi thought to be either embolic, formed in situ, or both [45]. Moreover, activated platelets, neutrophils, endothelial cells, neutrophil extracellular traps, microparticles, and coagulation proteases in ARDS have been associated with the process of deep vein thrombosis formation [46]. This explains the high prevalence of acute pulmonary embolism recently reported in COVID-19 patients admitted for hypoxemic acute respiratory failure [47]. Accumulating evidence suggests that SARS-CoV-2 might induce a cytokine storm (Figure 2) [48]. Patients with severe pneumonia or ARDS develop systemic manifestations of hyper inflammation called secondary hemophagocytic lymphocytosis (sHLH), also called macrophage activation syndrome (MAS) [49]. This hyperinflammation is characterized by an increased release of cytokines, particularly interleukin (IL)-6, IL-1 by binding to ACE-2 in the lung tissue, IL-6, interferon-γ and tumor necrosis factor-α [50]. This exaggerated response is responsible for significant tissue damage that could lead to multiorgan failure. Multiple studies comparing severe and moderate forms of COVID-19 concluded that the cytokine storm is associated with the severity of the disease [51,52].
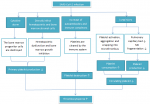
Many studies reported thrombocytopenia in patients with COVID-19, but its mechanism is still unclear. Xu et al. [53] described the hematological changes of thrombocytopenia in patients with COVID-19 and proposed three possible mechanisms (Figure 3) [53] by which SARS-CoV-2 can induce thrombocytopenia: a) SARS-CoV-2 can reduce platelet production by direct infection of bone marrow cells and therefore inhibit platelet synthesis. The virus can also do so through the cytokine storm that destroys bone marrow progenitor cells; b) COVID-19 infection can increase autoantibodies and immune complexes resulting in the specific destruction of platelets by the immune system; c) and damages of the lung tissues and pulmonary endothelial cells by the SARS-CoV-2 infection activate platelets in the lungs, resulting in aggregation and formation of microthrombi, which leads to an increase in platelet consumption.
Cardiovascular involvement: the cardiovascular (CV) system can be involved in different ways. The common mechanisms responsible for cardiovascular complications in COVID-19 are: a) direct myocardial injury [54]: SARS-CoV-2 enters human cells by binding to angiotensin-converting enzyme 2 (ACE2), a membrane-bound aminopeptidase that is highly expressed in the heart and lungs. ACE2 plays an important role in the neurohumoral regulation of the CV system under normal health conditions as well as in various diseases. Binding between the SARS-CoV-2 and ACE2 can lead to alterations in ACE2 signaling pathways, leading to acute damage to the myocardium and lungs; b) systemic inflammation [54]: the most severe forms of COVID-19 are characterized by an acute systemic inflammatory response and cytokine storm, which can lead to multi-organ injury, leading to organ failure. Studies have shown elevated circular levels of pro-inflammatory cytokines in patients with critical COVID-19; c) altered myocardial supply-demand relationship [55]: the increased metabolic demand associated with systemic infection coupled with hypoxia caused by the acute respiratory disease may alter the relationship between myocardial oxygen supply and demand and result in acute myocardial injury; d) plaque rupture and coronary thrombosis [56]: systemic inflammation and increased shear stress due to increased coronary blood flow may precipitate plaque rupture and result in acute myocardial infarction. The prothrombotic appearance created by systemic inflammation further increases the risk; e) adverse effects of various therapies [57]: various antiviral drugs, corticosteroids, and other therapies to treat the COVID-19 channel also have deleterious effects on the CV system. Electrolyte imbalances - electrolyte imbalances can occur in any critical systemic disease and precipitate arrhythmias, particularly in hospitalized patients with underlying heart disease. Of particular concern is the hypokalemia of COVID-19 due to the interaction of SARS-CoV-2 with the renin-angiotensin-aldosterone system, which increases susceptibility to various cardiac arrhythmias.
Role of underlying CV co-morbidities patients with the pre-existing cardiovascular disease appear to have an increased susceptibility to the development of COVID-19 and tend to have more severe disease with poorer clinical outcomes. Although the prevalence of diabetes and hypertension in this cohort is the same as in the general Chinese population, the prevalence of the cardiovascular disease is significantly higher. More importantly, the presence of diabetes, CVD and hypertension were associated with a two-fold, three-fold and two-fold increased risk of serious illness or requiring admission to an intensive care unit (ICU), suggesting a prognostic impact of these co-morbidities [57].
Acute kidney injury: the acute kidney injury (AKI) is an independent mortality factor, in SARS-CoV-2 infection, with an OR of 4.057 (95% CI 1.46-11.27; p<0.001) [58]. SARS-CoV-2-related AKI is a serious complication in 0.5 to 29% of hospitalized patients, particularly in the ICU setting [59-62], due to various etiology such as hemodynamic and cardiac disorders, invasive mechanical ventilation with impaired gas exchange, and occurrence of nosocomial sepsis; in addition to inflammatory substances release, microcirculatory alteration, and tubular injury [63]. Other factors are risks for AKI in acute respiratory distress syndrome (ARDS) including age, the severity of the illness, diabetes, obesity, and the history of heart failure [64]. Furthermore, the abundance, in the kidney tubular cells, of ACE2 as receptor facilitates the entry into target cells [26,27]; this infection participates in the worsening of the local inflammation, thus the incidence and the duration of AKI episodes [65]. In a postmortem renal histopathological analysis, Su et al. noted the invasion of SARS-CoV-2 into kidney parenchyma with diffuse proximal tubule injury, non-isometric vacuolar degeneration and even frank necrosis was observed, prominent erythrocyte aggregates obstructing the lumen of capillaries without platelet or fibrinoid material, without vasculitis, interstitial inflammation or hemorrhage, by light microscopy. The electron microscopic study described clusters of coronavirus in the tubular epithelium and podocytes. Additionally, ACE2 receptors were upregulated, and immunostaining with SARS-CoV-2 nucleoprotein antibody was positive in tubules [66]. Summarily, three mechanisms are involved in the SARS-CoV-2 associated AKI: cytokine damage, lung, and heart interactions and systemic effects [67] without forgetting direct lesions of the virus.
Neurological involvement: in the immunocompetent host, viral brain infections are rare, in general. But, neurotropic viruses can infect the central nervous system (CNS) (encephalitis, meningitis, myelitis, strokes) in the presence of temporary immunosuppression. The peripheral nervous system can be also affected (neuropathy, Guillain-Barré syndrome) [68]. The neurological complications during epidemic SARS-CoV-2 were not well studied. However, some case reports were presented with axonal peripheral neuropathy, myopathy with elevated creatinine kinase, or stroke [69-71]. For COVID-19, neurologic presentations were 36.4% of cases, more declared in severe patients (45.5% vs. 30.2% in non-severe patients). They can be subdivided into three groups: central nervous system manifestations (dizziness, headache, coma, acute cerebrovascular disease (intracerebral hemorrhage, hemorrhagic necrotizing encephalopathy, ischemic stroke), ataxia, acute myelitis, and seizure), peripheral nervous system manifestations (taste impairment, smell impairment, vision impairment, cranial neuropathies, Guillain-Barré Syndrome, and nerve pain) and skeletal muscle injury manifestations [72-78].
It is known that SARS-CoV-2 can induce neurological disease [79]. Effectively, Xu et al. mentioned the evident presence of SARS-CoV-2 in the brain of infected patients from the culture of the brain suspension [80]. Moreover, ACE2 receptors exist in the brainstem and the regions responsible for the regulation of cardiovascular function and were found in both neurons and glia [81,82]. Additionally, Yu et al. showed that SARS-CoV-2 belongs to the beta-coronavirus subgroup including MERS-CoV and SARS-CoV and has a high homological sequence with SARS-CoV-2 in the genomic analysis [83]. Given that, SARS-CoV-2 could have a potential neuroinvasion [84].
Regarding the route of invasion, first, the hematological and the lymphatic appear to be implausible because of the absence of virus particles in the non-neuronal cells in the infected brain areas [80,85,86]. Second, the synapse-connected route is more pointed out as possible via peripheral nerve terminals [87-90]. Third, since the last route has been described in other CoVs and avian bronchitis virus [87,89-92], there are case reports about smell or taste disturbances in the early stage of the disease [72]. Fourth, systemic inflammatory storm damaged the blood-brain-barrier and therefore sustained neuroinflammation [93].
Hepatic damage: Huang et al. [35] announced that 62% of critically ill patients had elevated aspartate aminotransferase (AST). Moreover, in a cohort of 1099 patients, Guan et al. observed that severe patients had more abnormal aminotransferase levels compared to non-severe [59]. In patients with COVID-19, the virus can directly affect the liver because about 2-10% of patients with COVID-19 develop diarrhea with SARS-CoV-2 RNA in stool and blood [94]. Liver disease is likely to be multifactorial and requires close biological monitoring to anticipate any progression. First, given that ACE2 receptors are expressed in cholangiocytes, SARS-CoV-2 may fix directly to cholangiocytes [95] and causes abnormal liver tests. Second, immune-mediated inflammation such as cytokine storm and hypoxia may also contribute to liver damage in critically ill patients with COVID-19. Third, there is a possible role of hepatotoxic drugs, especially lopinavir and ritonavir. Fourth, ventilation of severe recruitable ARDS with high levels of positive expiratory pressure may contribute to hepatic congestion [96]. Finally, in the histological examination, Cai et al. showed vesicular steatosis and degeneration of hepatocytes in the interlobular region and the hepatic sinuses and inflammatory cells in the hepatic sinuses [97]. However, no viral inclusions have been detected [98].
COVID-19 is a “great imitator”. It primarily infects the lung and may invade the cardiovascular system, central and peripheral nervous systems, kidneys, and liver. Thus, the management is multidisciplinary including ventilator and hemodynamic supports, pharmacologic treatment, and specific technics such as plasma exchange, stem cells convalescent plasma.
What is known about this topic
- SARS-CoV-2 is an emerging infection;
- SARS-CoV-2 affects essentially the pulmonary tract;
- Other organs might be impaired.
What this study adds
- SARS-CoV-2 could cause multiorgan impairment;
- The initial presentation is sometimes unusual;
- The multiorgan involvement requires multidisciplinary management.
The authors declare no competing interests.
AH, HR and HE participated in the conceptualization of the study, sourced the materials, drafted the manuscript, read and approved the final version of the manuscript.
Figure 1: potential therapeutic targets for SARS-CoV-2 and COVID-19
Figure 2: hyper-cytokinemia accompanying the overlap between acute respiratory distress syndrome (ARDS) and macrophage activation syndrome (MAS) associated with COVID-19
Figure 3: the possible mechanisms of thrombocytopenia in COVID-19 patients
- Tyrrell DA, Bynoe ML. Cultivation of a novel type of common-cold virus in organ cultures. Br Med J. 1965;1(5448):1467-1470. PubMed | Google Scholar
- Kin N, Miszczak F, Lin W, Gouilh MA, Vabret A, EPICOREM Consortium. Genomic analysis of 15 human coronaviruses OC43 (HCoV-OC43s) circulating in France from 2001 to 2013 reveals a high intra-specific diversity with new recombinant genotypes. Viruses. 2015;7(5):2358-2377. PubMed | Google Scholar
- Wevers BA, van der Hoek L. Recently discovered human coronaviruses. Clin Lab Med. 2009;29(4):715-724. PubMed | Google Scholar
- Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319-1325. PubMed | Google Scholar
- World Health Organization. Severe acute respiratory syndrome (SARS). 2003. Google Scholar
- Raj VS, Osterhaus AD, Fouchier RA, Haagmans BL. MERS: emergence of a novel human coronavirus. Curr Opin Virol. 2014 April;5:58-62. PubMed | Google Scholar
- Worldometers. COVID-19 coronavirus pandemic. Accessed 4th May 2020.
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536-544. PubMed | Google Scholar
- Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA et al. Severe acute respiratory syndrome-related coronavirus: The species and its viruses-a statement of the coronavirus study group. bioRxiv. 2020. Google Scholar
- World Health Organization (WHO). Pneumonia of unknown cause - China. 2020.
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221-236. PubMed | Google Scholar
- Hui DS, I Azhar E, Madani TA, Ntoumi F, Kock R, Dar O et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health-the latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020 Feb;91:264-266. PubMed | Google Scholar
- Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-273. PubMed | Google Scholar
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-269. PubMed | Google Scholar
- Pathak N. The great invader: how COVID-19 attacks every organ. Medscape. 2020. Google Scholar
- Dong X, Cao YY, Lu XX, Zhang JJ, Du H, Yan YQ et al. Eleven faces of coronavirus disease 2019. Allergy. 2020;75(7):1699-1709. PubMed | Google Scholar
- New England Journal of Medicine database. Coronavirus (COVID-19). 2020.
- European Society of Intensive Care Medicine. Coronavirus - public health emergency. 2020
- Novel Coronavirus Information Center. Elsevier Information Center. 2020.
- Novel coronavirus (COVID-19) news and resources. Medscape center. 2020.
- Gralinski LE, Menachery VD, Morgan AP, Totura AL, Beall A, Kocher J et al. Allelic variation in the toll-like receptor adaptor protein Ticam2 contributes to SARS-coronavirus pathogenesis in mice. G3 (Bethesda). 2017;7(6):1653-1663. PubMed | Google Scholar
- Zhao J, Yang Y, Huang H, Li D, Gu D, Lu X et al. Relationship between the ABO blood group and the COVID-19 susceptibility. Clin Infect Dis. 2020 Aug 4:ciaa1150. PubMed | Google Scholar
- Cheng Y, Cheng G, Chui CH, Lau FY, Chan PK, Ng MH et al. ABO blood group and susceptibility to severe acute respiratory syndrome. JAMA. 2005;293(12):1450-1451. PubMed | Google Scholar
- Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A et al. Human leukocyte antigen susceptibility map for SARS-CoV-2. J Virol. 2020;94(13):e00510-20. PubMed
- Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med Genet. 2003 Sep 12;4:9. PubMed | Google Scholar
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450-454. PubMed | Google Scholar
- Hamming I, Timens W, Bulthuis MLC, Lely AT, GJ Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: a first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631-637. PubMed | Google Scholar
- Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al. A crucial role of angiotensin-converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875-879. PubMed | Google Scholar
- Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444-1448. PubMed | Google Scholar
- Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586-590. PubMed | Google Scholar
- Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127-20. PubMed | Google Scholar
- The COVID-19 host genetics initiative.
- COVID-19: several imagine teams mobilize. Institut Imagine. 2020.
- Misra DP, Agarwal V, Gasparyan AY, Zimba O. Rheumatologists' perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol. 2020 Jul;39(7):2055-2062. PubMed | Google Scholar
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497-506. PubMed | Google Scholar
- Sun P, Qie S, Liu Z, Ren J, Xi JJ. Clinical characteristics of 50466 patients with 2019-nCoV infection. medRxiv. 2020. Google Scholar
- Tian S, Hu W, Niu L, Liu H, Xu H, Xiao S-Y. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700-704. PubMed | Google Scholar
- ARDS Definition Task Force, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526-2533. PubMed | Google Scholar
- Aublanc M, Perinel S, Guérin C. Acute respiratory distress syndrome mimics: the role of lung biopsy. Curr Opin Crit Care. 2017;23(1):24-29. PubMed | Google Scholar
- Gattinoni L, Chiumello D, Rossi S. COVID-19 pneumonia: ARDS or not. Crit Care. 2020;24(1):154. PubMed | Google Scholar
- Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L et al. COVID-19 pneumonia: different respiratory treatments for different phenotypes. Intensive Care Med. 2020;46(6):1099-1102. PubMed | Google Scholar
- Gattinoni L, Caironi P, Cressoni M, Chiumello D, Ranieri VM, Quintel M et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006;354(17):1775-1786. PubMed | Google Scholar
- Cressoni M, Cadringher P, Chiurazzi C, Amini M, Gallazzi E, Marino A et al. Lung inhomogeneity in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2014;189(2):149-158. PubMed | Google Scholar
- Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364-374. PubMed | Google Scholar
- Tomashefski JF, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983;112(1):112-126. PubMed | Google Scholar
- Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration. 2017;93(3):212-225. PubMed | Google Scholar
- Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46(6):1089-1098. PubMed | Google Scholar
- Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934-943. PubMed | Google Scholar
- Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033-1034. PubMed | Google Scholar
- Tveiten H, Aukrust P, Lehne G, Rodriguez JR, Skjønsberg OH. Haemophagocytic lymphohistiocytosis in COVID-19 cases? Tidsskr Nor Laegeforen. 2020;140(6). PubMed | Google Scholar
- Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620-269. PubMed | Google Scholar
- Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473-474. PubMed | Google Scholar
- Xu P, Zhou Q, Xu J. Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol. 2020;99(6):1205-1208. PubMed | Google Scholar
- Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020 March;46(5):846-848. PubMed | Google Scholar
- Chen C, Chen C, Yan JT, Zhou N, Zhao JP, Wang DW. Analysis of myocardial in- jury in patients with COVID-19 and association between concomitant cardiovascular diseases and severity of COVID-19. Zhonghua Xin Xue Guan Bing Za Zhi. 2020 Mar 6;48(7):567-571. PubMed | Google Scholar
- Warren-Gash C, Hayward AC, Hemingway H, Denaxas S, Thomas SL, Timmis AD et al. Influenza infection and risk of acute myocardial infarction in England and Wales: a caliber self-controlled case series study. J Infect Dis. 2012;206(11):1652-1659. PubMed | Google Scholar
- Guo T, Fan Y, Chen M, Wu X, Zhang L, He T et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5(7):811-818. PubMed | Google Scholar
- Chu KH, Tsang WK, Tang CS, Lam MF, Lai FM, To KF et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005;67(2):698-705. PubMed | Google Scholar
- Guan W, Ni Z, Hu Y, Liang W, Ou C, He J et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708-1720. PubMed | Google Scholar
- Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054-1062. PubMed | Google Scholar
- Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW et al. Presenting characteristics, comorbidities and outcomes among 5700 patients hospitalized with COVID-19 in the NewYork City area. JAMA. 2020;323(20):2052-2059. PubMed | Google Scholar
- Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centred, retrospective, observational study. Lancet Respir Med. 2020;8(5):475-481. PubMed | Google Scholar
- Joannidis M, Forni LG, Klein SJ, Honore PM, Kashani K, Ostermann M et al. Lung-kidney interactions in critically ill patients: consensus report of the Acute Disease Quality Initiative (ADQI) 21 Workgroup. Intensive Care Med. 2020;46(4):654-672. PubMed | Google Scholar
- KDIGO Working Group. Clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2(1):2.
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B beta coronaviruses. Nat Microbiol. 2020;5(4):562-569. PubMed | Google Scholar
- Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98(1):219-227. PubMed | Google Scholar
- Ronco C, Reis T. Kidney involvement in COVID-19 and rationale for extracorporeal therapies. Nat Rev Nephrol. 2020;16(6):308-310. PubMed | Google Scholar
- Berth SH, Leopold PL, Morfini GN. Virus-induced neuronal dysfunction and degeneration. Front Biosci (Landmark Ed). 2009 Jun 1;14:5239-5259. PubMed | Google Scholar
- Tsai LK, Hsieh ST, Chao CC, Chen YC, Lin YH, Chang SC et al. Neuromuscular disorders in severe acute respiratory syndrome. Arch Neurol. 2004;61(11):1669-1673. PubMed | Google Scholar
- Ding Y, Wang H, Shen H, Li Z, Geng J, Han H et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200(3):282-289. PubMed | Google Scholar
- Hwang CS. Olfactory neuropathy in severe acute respiratory syndrome: report of a case. Acta Neurol Taiwan. 2006;15(1):26-28. PubMed | Google Scholar
- Mao L, Jin H, Wang M, Hu Y, Chen S, He Q et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):683-690. PubMed | Google Scholar
- Zhao K, Huang J, Dai D, Feng Y, Liu L, Nie S. Acute myelitis after SARS-CoV-2 infection: a case report. medRxiv. 2020.03.16.20035105. Google Scholar
- Sharifi-Razavi A, Karimi N, Rouhani N. COVID-19 and intracerebral hemorrhage: causative or coincidental. New Microbes and New Infect. 2020 Mar 27;35:100669. PubMed | Google Scholar
- Dinkin M, Gao V, Kahan J, Bobker S, Simonetto M, Wechsler P et al. COVID-19 presenting with ophthalmoparesis from cranial nerve palsy. Neurology. 2020;95(5):221-223. PubMed | Google Scholar
- Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B. COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology. 2020;296(2):E119-E120. PubMed | Google Scholar
- Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG et al. Guillain-Barré syndrome associated with SARS-CoV-2. N Engl J Med. 2020;382(26):2574-2576. PubMed | Google Scholar
- Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 2020;382(23):2268-2270. PubMed | Google Scholar
- Glass WG, Subbarao K, Murphy B, Murphy PM. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV-2) pulmonary infection of mice. J Immunol. 2004;173(6):4030-4039. PubMed | Google Scholar
- Xu J, Zhong S, Liu J, Li L, Li Y, Wu X et al. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis. 2005;41(8):1089-1096. PubMed | Google Scholar
- Gowrisankar YV, Clark MA. Angiotensin II regulation of angiotensin-converting enzymes in spontaneously hypertensive rat primary astrocyte cultures. J Neurochem. 2016;138(1):74-85. PubMed | Google Scholar
- Xia H, Lazartigues E. Angiotensin-converting enzyme 2: central regulator for cardiovascular function. Curr Hypertens Rep. 2010;12(3):170-175. PubMed | Google Scholar
- Yu F, Du L, Ojcius DM, Pan C, Jiang S. Measures for diagnosing and treating infections by a novel coronavirus responsible for a pneumonia outbreak originating in Wuhan, China. Microbes Infect. 2020;22(2):74-79. PubMed | Google Scholar
- Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV-2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. 2020;92(6):552-555. PubMed | Google Scholar
- Ding Y, He L, Zhang Q, Huang Z, Che X, Hou J et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV-2) in SARS patients: implications for pathogenesis and virus transmission pathways. J Pathol. 2004;203(2):622-630. PubMed | Google Scholar
- Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y et al. Multiple organ infection and the pathogenesis of SARS. J Exp Med. 2005;202(3):415-424. PubMed | Google Scholar
- Li YC, Bai WZ, Hirano N, Hayashida T, Hashikawa T. Coronavirus infection of rat dorsal root ganglia: ultrastructural characterization of viral replication, transfer and the early response of satellite cells. Virus Res. 2012;163(2):628-635. PubMed | Google Scholar
- Li YC, Bai WZ, Hirano N, Hayashida T, Taniguchi T, Sugita Y et al. Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol. 2013;521(1):203-212. PubMed | Google Scholar
- Andries K, Pensaert MB. Immunofluorescence studies on the pathogenesis of hemagglutinating encephalomyelitis virus infection in pigs after oronasal inoculation. Am J Vet Res. 1980;41(9):1372-1378. PubMed | Google Scholar
- Matsuda K, Park CH, Sunden Y, Kimura T, Ochiai K, Kida H et al. The vagus nerve is one route of transneuronal invasion for intranasally inoculated influenza a virus in mice. Vet Pathol. 2004;41(2):101-107. PubMed | Google Scholar
- Mengeling WL, Boothe AD, Ritchie AE. Characteristics of a coronavirus (strain 67N) of pigs. Am J Vet Res. 1972;33(2):297-308. PubMed | Google Scholar
- Chasey D, Alexander DJ. Morphogenesis of avian infectious bronchitis virus in primary chick kidney cells. Arch Virol. 1976;52(1-2):101-111. PubMed | Google Scholar
- Sankowski R, Mader S, Valdes-Ferrer SI. Systemic inflammation and the brain: novel roles of genetic, molecular and environmental cues as drivers of neurodegeneration. Front Cell Neurosci. 2015 Feb 2;9:28. PubMed | Google Scholar
- Yeo C, Kaushal S, Yeo D. Enteric involvement of coronaviruses: is fecal-oral transmission of SARS-CoV-2 possible. Lancet Gastroenterol Hepatol. 2020;5(4):335-337. PubMed | Google Scholar
- Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A et al. Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. bioRxiv. 2020. Google Scholar
- Zhang C, Shi L, Wang FS. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5(5):428-430. PubMed | Google Scholar
- Cai Q, Huang D, Yu H, Zhu Z, Xia Z, Su Y et al. COVID-19: abnormal liver function tests. J Hepatol. 2020;73(3):566-574. PubMed | Google Scholar
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420-422. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
