
A case report of Arnold Chiari type 1 malformation in acromesomelic dwarf infant
Miteshkumar Rajaram Maurya, Renju Ravi, Sona Ajit Pungavkar
Corresponding author: Miteshkumar Rajaram Maurya, Department of Clinical Pharmacology, Seth Gordhandas Sunderdas Medical College and King Edward Memorial Hospital, Mumbai, Maharashtra, India 
Received: 04 Dec 2020 - Accepted: 03 Jan 2021 - Published: 18 Jan 2021
Domain: Neurodevelopmental Disabilities,Neuroradiology,Pediatric neurology
Keywords: Arnold Chiari 1 malformation, acromesomelic dwarfism, hydrocephalus, neural tube defects, case report
©Miteshkumar Rajaram Maurya et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Miteshkumar Rajaram Maurya et al. A case report of Arnold Chiari type 1 malformation in acromesomelic dwarf infant. Pan African Medical Journal. 2021;38:58. [doi: 10.11604/pamj.2021.38.58.27295]
Available online at: https://www.panafrican-med-journal.com//content/article/38/58/full
Case report 

A case report of Arnold Chiari type 1 malformation in acromesomelic dwarf infant
A case report of Arnold Chiari type 1 malformation in acromesomelic dwarf infant
Miteshkumar Rajaram Maurya1,&, Renju Ravi1, Sona Ajit Pungavkar2
&Corresponding author
Arnold Chiari malformation is one of the commonest cause of congenital hydrocephalus. Cause of fetal development of cerebellar tonsils remains unknown and may be diagnosed at later in life. The association of Arnold Chiari malformation with acromesomelic dwarfism is not known. We report male infant diagnosed with acromesomelic dwarfism at end of gestation period on basis of antenatal ultrasonography findings. An ultrasound scan of infant head at fifth month of birth was performed in view of increasing head circumference that revealed aqueductal stenosis with dilated posterior horn of lateral ventricles in brain.

Arnold Chiari malformations were first described in pediatric autopsy specimen in 1891 by Hans Chiari, an Austrian Pathologist (1851-1916) [1]. In legacy with name of his professor Dr. Arnold and his name Hans Chiari, the hind brain disorder is named as Arnold Chiari malformation [2]. Exact etiology remains unknown. Arnold Chiari type I malformation remains mostly asymptomatic until adulthood. Diagnosis is made by measuring the length of protrusion of cerebellar tonsils below the inner margins of foramen magnum to the inferior most part of tonsils (measurement taken from ophisthion to basion). If tonsils are above the foramen magnum considered as normal, if tonsillar length is <5mm is also normal but the term benign tonsillar ectopia can be used and if tonsillar length is >5 mm then it is termed as Arnold Chiari type I malformation [3]. Incidence of Arnold Chiari malformation is gradually increasing because of increased detection with various imaging techniques available. Hydrocephalus is one of the common presentation of Arnold Chiari 1 malformation.

A 5-month-old baby weighing 2.6 kg at birth and born of non-consanguineous marriage presented to the pediatrician with increase in the head circumference. The child was feeding well and there were no other complaints. At the ninth month of pregnancy, the child has been diagnosed with acromesomelic dwarfism during a routine antenatal scan. The mother gave a history of the first child having severe hydrocephalus with undeveloped crania (on intrauterine sonogram) at 19 weeks of gestation. The pregnancy was terminated. The mother was subsequently put on the multi-vitamin and folic acid tablet supplementation before the second conception. The second conception occurred 2.5 years later.
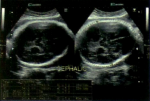
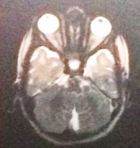
An ultrasound scan of head done at fifth month of birth (Figure 1) showed ventriculomegaly with dilatation of posterior horn (transverse diameter of 2.4cm). The third and lateral ventricles appeared dilated (anterior horn of lateral ventricle on left side measured 23mm in width and the posterior horn measured-24mm). The fourth ventricles were normal. No intra-cranial or intra-ventricular/germinal matrix hematoma was detected. The brain parenchyma was normal as were the sulci and gyri and frontal, temporal and parietal lobes. No subdural or extradural collection was seen. A provisional diagnosis was made of infantile hydrocephalus or aqueductal stenosis and a MRI brain with screening of whole spine was advised by pediatrician. MRI scan brain with screening of whole spine was done at the fifth month of birth that revealed herniation of cerebellum tonsil (4mm) through the foramen magnum into the upper cervical canal consistent with Arnold Chiari type 1 malformation with no evidence of syrinx formation or cord edema (Figure 2 and Figure 3). There was compression of the cervico-medullary junction with mild to moderate obstructive hydrocephalus, without periventricular ooze. There was atrophy of both lentiform nuclei with paucity of white matter in bilateral occipital regions with thinning of the corpus callosum. There was rounded 3 mm distended subarachnoid spaces around the optic nerves. Cerebral sulci well seen. Aqueduct canal was well formed. Evidence of occluded subarachnoid spaces at foramen magnum was present. There was no evidence of neural tube defects.
Next generation sequencing (NGS) (Table 1) revealed presence of heterozygous LAMB1 (encodes for laminin subunit beta-1 protein) and MPDZ gene (encodes for Multiple PDZ domain protein) mutation from paternal side whereas that of GAA gene (encodes for acid alpha-glucosidase also known as acid maltase synthesized in lysosomes) mutation from maternal side (gene associated with congenital hydrocephalus). LAMB1, MPDZ were analyzed because the mutations in either are known to be associated with congenital hydrocephalus with autosomal recessive genetic inheritance. The mutation in fibroblast growth factor receptor 3 (FGFR3) gene responsible for rhizomelic short stature was present in infant producing clinical picture of achondroplasia, which is autosomal dominant genetic condition. Definitive prenatal diagnosis can be offered at present only for achondroplasia gene (FGFR3) mutation, the empiric recurrence risk being around 5% (to account for possibility of germinal mosaicism). A neurosurgery consultation was done and ventriculo-peritoneal shunt or third ventriculostomy or suboccipital craniectomy with first and second vertebra laminectomy was advised. However, the guardian of patient were reluctant to go for this procedure due to unsurity of benefits and repeat surgery need as explained to them.
This paper describes a case of an infant presenting with progressive increase in head circumference at fifth month of gestation and diagnosed with Arnold Chiari malformation type 1. There are only isolated reports of Arnold Chiari type 1 malformation or acromesomelic dwarfism in the Indian population. Very few cases present with Arnold Chiari type 1 malformation in association with acromesomelic dwarfism and progression of hydrocephalus [4-7]. Danda S et al. study reported about a family siblings affected with acromesomelic dwarfism [4]. Haldar et al. described about the anesthesia concerns involved in patients with acromesomelic dysplasia with associated hydrocephalus, Arnold Chiari malformation and syringomyelia [5]. Arnold Chiari malformation is almost always associated with neural tube defects [6].
The exact etiology of Arnold Chiari malformation and acromesomelic short stature remains unknown. A large metanalysis strongly suggests that the MTHFD 1 G1958A gene polymorphism is strongly linked with neural tube defect [7]. Urbizu et al. identified four genetic variants (located in the genes ALDH1A2, CDX1 and FLT1) to be associated with adult classic Chiari malformation type 1 [8]. Markunas et al. identified different levels of expression in genes related with dorso-ventral axis formation (ETS1, ETS2, NOTCH4), ribosome, spliceosome and proteasome in pediatric classic Chiari malformation type 1 patients [9]. There is limited information on MTHFR (methylene tetrahydrofolate reductase) gene mutation prevalent among the Indian population that may interfere with benefit of folic acid supplementation. Methylene Tetrahydrofolate Dehydrogenase gene (MTHFD) is also one of the key genes involved in folate metabolism pathway.
The fundamental challenge lies in early radiological diagnosis and detection of cases of dwarfism (acromesomelic type) as well as Arnold Chiari 1 malformation. Also there exist lack of awareness about the branch of fetal medicine and perinatalogists who have expertise diagnosing the fetal malformations doing genetic workup for the same. Further evaluation need to be done for the pathophysiology of this disorder (viral origin/drug induced/deficiency disorders/environmental factors/genetics) facilitating early prevention and appropriate intervention.
Our study presents lately diagnosed case of Arnold Chiari I malformation in an infant after birth. However, we do not know if there is any association between acromesomelic dwarfism and Arnold Chiari malformation. There is need to create awareness among the people about the importance of early antenatal anomaly scan and genetic screening with emerging branch of fetal medicine experts to diagnose early the occurrence of such malformations.
The authors declare no competing interests.
Miteshkumar Rajaram Maurya: patient case evaluation, data and report collection, drafting manuscript; Renju Ravi: patient case evaluation, data and report collection, drafting manuscript; Sona Ajit Pungavkar: diagnosing the case by radio-diagnostic imaging, patient case evaluation, providing the report, drafting manuscript and corrections. All the authors have read and agreed to the final manuscript.
Table 1: genetic work up for infant diagnosed with rhizomelic short stature and hydrocephalus; infant was diagnosed with heterozygous mutation in following genes in next generation sequencing (NGS) for FGFR3, MPDZ, LAMB1, GAA gene
Figure 1: antenatal ultrasound scan of brain
Figure 2: magnetic resonance imaging (MRI) of the brain
Figure 3: magnetic resonance imaging (MRI) scan screening of whole spine
- Tubbs RS, Cohen-Gadol AA. Cohen-Gadol Hans Chiari (1851-1916). J Neurol. 2010;257(7):1218-20. PubMed | Google Scholar
- Kumar S, Sarmah BJ, Bora P. Arnold Chiarri malformation type II. Ped Rad (serial online). 2009;9(10).
- Elster AD, Chen MY. Chiari I malformation: clinical and radiological reappraisal. Radiology. 1992;183(2):347-53. PubMed | Google Scholar
- Danda S, Phadke SR, Agarwal SS. Acromesomelic dwarfism: report of a family with two affected siblings. Indian Pediatr. 1997 Dec;34(12):1127-30. PubMed
- Haldar R, Gyanesh P, Samanta S. Anesthesia for a patient of acromesomelic dysplasia with associated hydrocephalus, Arnold Chiari malformation and syringomyelia. J Anaesthesiol Clin Pharmacol. 2013 Oct;29(4):555-7. PubMed | Google Scholar
- Ganesh D, Sagayaraj BM, Barua RK, Sharma N, Ranga U. Arnold Chiari malformation with spina bifida: a lost opportunity of folic Acid supplementation. J Clin Diagn Res. 2014 Dec;8(12):OD01-3. PubMed | Google Scholar
- Jiang J, Zhang Y, Wei L, Sun Z, Liu Z. Association between MTHFD1 G1958A polymorphism and neural tube defects susceptibility: a meta-analysis. PLoS One. 2014 Jun 30;9(6):e101169. PubMed | Google Scholar
- Urbizu A, Khan TN, Ashley-Koch AE. Genetic dissection of Chiari malformation type 1 using endophenotypes and stratification. J Rare Dis Res Treat. 2017;2(2):35-42. Google Scholar
- Markunas CA, Lock E, Soldano K, Cope H, Ding CK, Enterline DS et al. Identification of Chiari type I malformation subtypes using whole genome expression profiles and cranial base morphometrics. BMC medical genomics. 2014;7:39. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
Recently from the PAMJ








Authors´ services
