
Right ventricular dysplasia in the elderly: a case report from autopsy
Alban Ikenna Mgbehoma, Oluwaseye Olumide Onayemi, Sunday Sokunle Soyemi, John Oladapo Obafunwa
Corresponding author: Alban Ikenna Mgbehoma, Department of Pathology and Forensic Medicine, Lagos State University Teaching Hospital, Ikeja, Lagos, Nigeria 
Received: 08 Apr 2021 - Accepted: 14 Apr 2021 - Published: 28 Apr 2021
Domain: Forensic Medicine
Keywords: Right ventricular cardiomyopathy, right ventricular dysplasia, case report
©Alban Ikenna Mgbehoma et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Alban Ikenna Mgbehoma et al. Right ventricular dysplasia in the elderly: a case report from autopsy. Pan African Medical Journal. 2021;38:404. [doi: 10.11604/pamj.2021.38.404.29250]
Available online at: https://www.panafrican-med-journal.com//content/article/38/404/full
Case report 

Right ventricular dysplasia in the elderly: a case report from autopsy
Right ventricular dysplasia in the elderly: a case report from autopsy
![]() Alban Ikenna Mgbehoma1,&, Oluwaseye Olumide Onayemi1, Sunday Sokunle Soyemi1,
Alban Ikenna Mgbehoma1,&, Oluwaseye Olumide Onayemi1, Sunday Sokunle Soyemi1, ![]() John Oladapo Obafunwa1
John Oladapo Obafunwa1
&Corresponding author
Right ventricular dysplasia (RVD) is a rare disease of the heart that primarily affects the right ventricle. It is a clinical and pathological entity that presents classically with palpitations, syncope, or even sudden death. It presents rarely in the elderly. Where sudden death is the first and only presentation, an autopsy is required to make the diagnosis. However, the pathomorphological features of RVD can easily be overlooked or missed at autopsy. We report the case of a 68-year-old male with the past medical history of hypertension, gout and inflammatory bowel syndrome. He was admitted on account of difficulty in breathing, abdominal swelling and reduced urination. Physical examination revealed hypertension with cardiac murmurs, widespread crepitations, distended abdomen and lower limb oedema. Provisional diagnoses of acute-on-chronic kidney disease and congestive cardiac failure secondary to hypertensive heart disease, precipitated by probable gastrointestinal infection were made. While on admission, he had an episode of syncope. Electrocardiogram revealed bigeminy and bradycardic sinus rhythm with unifocal ventricular premature contraction. He died on the 8th week of admission. Autopsy revealed an enlarged heart weighing 600gm; there was thinning of the apical aspect of the right ventricular wall with subtotal fibrofatty replacement. Microscopic examination revealed a transmural replacement of cardiac myocytes by fibroadipose tissue extending inwards, in the most parts, from the epicardium to the endocardial surface. Our aim is to increase the awareness of these pathomorphological features among anatomic/forensic pathologists.

Right ventricular dysplasia (RVD) is a rare disease of the heart that primarily affects the right ventricular musculature [1]. This disease may impair the pump function of the heart or cause irregular heartbeats and these disturbances of normal heart function may culminate in sudden death [2]. It has been implicated as a cause of sudden death in the young, particularly athletes [3,4] and rarely manifests in the elderly [5]. Where sudden death is the clinical presentation, diagnosis can only be made if post mortem examination is conducted [6]. Making such a diagnosis at autopsy requires an established and functional medico-legal system that makes sudden and unexpected death a reportable case. However, this diagnosis can be easily missed or overlooked by the anatomic/forensic pathologists [7].
We report the case of a now deceased elderly male patient who had right ventricular dysplasia. A search of the Nigerian literature did not reveal any previous documentation of RVD. This report describes the pathomorphological features of RVD and reviews the literature. Our aim is to increase the awareness of these features among anatomic/forensic pathologists.

The now deceased was a 68-year-old male with a past medical history of hypertension, gout and inflammatory bowel syndrome. He was admitted on account of difficulty in breathing, abdominal swelling and reduced urination. Physical examination revealed hypertension with cardiac murmurs, widespread crepitations, distended abdomen and lower limb oedema. Provisional diagnoses of acute-on-chronic kidney disease and congestive cardiac failure secondary to hypertensive heart disease, precipitated by probable gastrointestinal infection were made. Laboratory evaluation showed azotaemia, hyponatraemia, hyperkalaemia, hyperphosphataemia, hyperuricaemia, elevated creatinine and elevated erythrocyte sedimentation rate (ESR). He was urgently dialysed. While on admission, he was managed for diarrhoea, bladder outlet obstruction and gouty arthritis; however, on the 20th day of admission, he had an episode of syncope. Following the syncopal episode, an electrocardiogram (ECG) was done and it revealed bigeminy and bradycardic sinus rhythm with unifocal ventricular premature contraction.
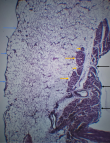
However, the urea and creatinine levels remained elevated and his consciousness diminished (Glasgow coma scale -7/15). He was again dialysed, re-evaluated and had 3 more sessions of dialysis. In addition, he was transfused with packed red blood cells on 3 different occasions. The patient´s clinical state progressively declined as evidenced by the persistent respiratory distress and diminished consciousness. He died on the 8th week of admission. An autopsy was performed upon the request of the family. External examination at autopsy revealed remarkable bilateral bony swelling at the knee joints and bilateral lower limb oedema. Internal examination revealed an enlarged heart weighing 600 gm. There was panvalvular dilatation with the tricuspid, pulmonary, mitral and aortic valves having circumferences of 13.0 cm, 9.0 cm, 9.5 cm, and 11.5 cm, respectively. The left and right ventricular wall thickness measured 2.0 cm and 0.6cm, respectively. However, at the distal third of the right ventricular free wall, that is, at the apical region, there was thinning of the wall (0.2 cm) with subtotal fibrofatty replacement of the myocardium (Figure 1). One of the papillary muscles on the left side had an external diameter of 2.0 cm. Other findings included bilateral pulmonary oedema, hepatomegaly, chronic passive hepatic congestion, multiple simple renal cysts with focal benign papillary cortical adenoma, benign prostatic hyperplasia, complicated abdominal aortic atherosclerosis and presence of many corpora amylacea in the cerebral cortex.
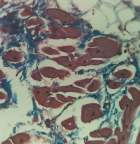
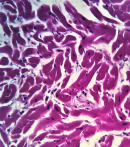
Microscopic examination of the sections of the heart taken from the apical region of the right ventricular wall revealed a transmural replacement of cardiac myocytes by fibroadipose tissue extending inwards, in the most parts, from the epicardium to the endocardial surface. Consequently, the myocardium showed at least 80-90% fat in-growth with focal transmural involvement (Figure 2). A Masson trichrome stain was done to highlight the fibrous tissue (Figure 3). Mild lymphocytic infiltrates were observed in parts (Figure 4). Sections taken from other parts of the right and left ventricular wall showed myocyte hypertrophy. The coronary vessels showed very mild eccentric atherosclerotic changes. Genetic studies were not done due to the unavailability of necessary facilities. Death was attributed to congestive cardiac failure due to hypertensive heart disease, with the right ventricular dysplasia being a significant and independent cause. The manner of death was concluded as natural.
The term right ventricular dysplasia (RVD) was coined by Fontaine in 1977 to describe a small group of patients who suffered from a strange heart disease characterised by ventricular tachycardia that originated from the right ventricle rather than from the usual left ventricular scar [8]. The more popular term “arrythmogenic” RVD (ARVD) was proposed because arrhythmia is the most frequent manifestation of this disease [9]. In the ensuing discussion, RVD and ARVD are used interchangeably. Thiene et al. in 1988, studied 60 hearts of young adults who had died from sudden cardiac death and observed RVD-like pathologies in 12 of them. They concluded that these individuals died from a cardiac muscle pathology of unknown aetiology, and named it right ventricular cardiomyopathy (RVC) [3]. In a later publication by this same group of researchers in 1996, the term arrhythmogenic right ventricular cardiomyopathy (ARVC) was introduced. That same year, the World Health Organisation (WHO) and the International Society and Federation of Cardiology (ISFC) task force on definition and classification of cardiomyopathies adopted the designation, ARVC, as the more appropriate name [10]. In other words, WHO and ISFC formally recognised ARVD as a cardiomyopathy and not a dysplasia; this partly de-emphasises suggestions of a congenital or growth abnormality. The ISFC task force went ahead to include ARVC as one of the five morphological and functional phenotypes of cardiomyopathies; the other four being dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM) and unclassified cardiomyopathy.
This development polarised the name of this disease entity amongst researchers: proponents of the “cardiomyopathy” terminology believe the disease to be an acquired condition, whereas, proponents of the “dysplasia” nomenclature believe that it is a developmental condition. Interestingly, Fontaine, who was the first to describe and name the disease, warned that “the term �cardiomyopathy´ may include too many different subtypes of diseased hearts with ARVD-like pathologies and render the search for a unified pathogenic mechanism and therapy very difficult, if not impossible” [11]. Right ventricular dysplasia presents classically with palpitations, syncope, or even sudden death [2]. The latter can be the first presenting symptom, particularly in young people under the age of 35 years and in athletes [3,4]. The incidence RVD is unknown; however, a prevalence of 1:1000, with male predominance has been reported [12]. Right ventricular dysplasia is familial with an autosomal dominant mode of inheritance [13]; its aetiopathogenesis is associated with mutation in the genes encoding any of the five major components of the cardiac desmosomes. These genes are: PKP2 (encoding plakophilin-2), DSG2 (encoding desmoglein-2), DSP (encoding desmoplakin), DSC2 (encoding desmocollin-2) and JUP (encoding junctional plakoglobin) [14].
Two pathological types have been described; the first is characterised solely by fat replacement of the right ventricle (fatty variant) while the second is characterised by fat infiltration and scarring of the right ventricle (fibrofatty variant). However, there is a controversy as to whether these types represent a spectrum of the same disease or whether they are different disease entities [15]. More controversies exist as regards the significance of myocardial fat infiltration. The fact that fat is present in the myocardium of normal hearts has posed a diagnostic challenge for anatomic/forensic pathologists at autopsy [16]. On the one hand, there is a tendency for the anatomic/forensic pathologists to regard intramyocardial fat as being physiological and underdiagnose RVD; or as pathological and over diagnose RVD. On the other hand, these features may as well go completely unnoticed. In other words, making the autopsy diagnosis of RVD could be problematic. The mere presence of adipose tissue, macroscopically, in the wall of the right ventricle is in itself insufficient to make the diagnosis; microscopic evaluation is pertinent. The microscopic examination of the retained sections revealed sheets of adipocytes interspersed by fibrous tissue strands. The latter is a more reliable feature for diagnosis [15,16]. The occurrence of myocyte atrophy with or without necrosis as well as the presence of lymphocyte infiltrates have also been reported [17,18]. Myocyte atrophy with mild lymphocyte infiltrate was observed in the index case (Figure 4). The fibrofatty infiltration occurs in the area of the heart referred to as the “triangle of dysplasia” [19]. The sides of the triangle are represented by the right ventricular outflow tract, the apex and the pulmonary infundibulum. In the present case, the fatty infiltration was in the apical region.
Another very important diagnostic feature, which has been reported in all cases, of RVD, is right ventricular (RV) wall thinning with or without aneurysmal dilatation. In the index case, the right ventricular wall thickness at the apical region measured 0.2 cm (reference interval = 0.3-0.5cm). Sometimes, there may be an isolated left ventricular or a biventricular involvement and where this is the case, the term arrhythmogenic cardiomyopathy (ACM) is employed [20]. To date, the growing body of literature on ARVD recognise RV wall thinning with fibrofatty replacement of the myocardium as the most consistent gross features, while myocyte atrophy with fat infiltration and fibrosis as the most reliable microscopic features.
Considering the potentially fatal outcome of RVD, a correct diagnosis made at autopsy would immediately initiate the screening cascade of the deceased´s relatives.
The authors declare no competing interests.
AIM: performed the autopsy, did literature search and prepared the draft manuscript; OOO: contributed to working up the case; SSS: contributed to literature review and review of the manuscript; JOO: reviewed the autopsy and the diagnosis, reviewed the manuscript and shared some literature material. All the authors have read and agreed to the final manuscript.
Figure 1: the right ventricular free wall showing ventricular wall thinning (green arrows) with marked fibrofatty replacement of the myocardium at the apex (black arrows), the papillary muscle (blue arrow) is also visible
Figure 2: histological section shows a focal transmural replacement of cardiac myocytes by fibroadipose tissue extending from the epicardium (blue arrows) to the endocardial surface (black arrows), an island of atrophic myocytes is seen floating in the fibroadipose matrix (yellow arrows), H&E X40
Figure 3: Masson trichrome - stained section of the heart highlighting the fibrous tissue (blue strands), X400
Figure 4: histological section shows myocyte atrophy with mild lymphocyte infiltrate (blue arrows), H&E X400
- Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry: study group on arrhythmogenic right ventricular dysplasia/cardiomyopathy of the working groups on myocardial and pericardial disease and arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation. 2000 Mar 21;101(11):E101-6. PubMed | Google Scholar
- Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005 Dec 20;112(25):3823-32. PubMed | Google Scholar
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988 Jan 21;318(3):129-33. PubMed | Google Scholar
- Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89(5):588-596. PubMed | Google Scholar
- Garcia-Quintana A, Ortega-Trujillo JR, Medina Fernandez-Aceytuno A. Arrhythmogenic right ventricular dysplasia in the elderly. Rev Esp Cardiol. 2007 Jan;60(1):87-9. PubMed | Google Scholar
- Basso C, Calabrese F, Corrado D, Thiene G. Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res. 2001;50(2):290-300. PubMed | Google Scholar
- Coumbe A, Perez-Martinez AL, Fegan AW, Hill IR. Arrhythmogenic right ventricular dysplasia (ARVD): an overlooked and underdiagnosed condition. Med Sci Law. 1997;37(3):262-265. PubMed | Google Scholar
- Fontaine G, Guiraudon G, Frank R, Vedel J, Grosgogeat Y, Cabrol C et al. Stimulation studies and epicardial mapping in ventricular tachycardia: study of mechanisms and selection for surgery. In - Reentrant Arrhythmias - HE Kulbertus Ed MTP Pub Lancaster: 1977;334-350.
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384-398. PubMed | Google Scholar
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J et al. Report of the 1995 World Health Organization/international society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation. 1996;93(5):841-842. PubMed | Google Scholar
- Fontaine G, Chen HS. Arrhythmogenic right ventricular dysplasia back in force. Am J Cardiol. 2014;113(10):1735-1739. PubMed | Google Scholar
- Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004 Dec;97(3):499-501. PubMed | Google Scholar
- Buja GF, Nava A, Martini B, Canciani B, Thiene G. Right ventricular dysplasia: a familial cardiomyopathy. Eur Heart J. 1989;10 Suppl D:13-15. PubMed | Google Scholar
- Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008 May;5(5):258-267. PubMed | Google Scholar
- Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases. Circulation. 1998;97(16):1571-1580. PubMed | Google Scholar
- Basso C, Thiene G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy: just a matter of fat. Cardiovasc Pathol. 2005;14(1):37-41. PubMed | Google Scholar
- Fontaliran F, Fontaine G, Brestescher C, Labrousse J, Vilde F. Significance of lymphoplasmocytic infiltration in arrhythmogenic right ventricular dysplasia: apropos of 3 own cases and review of the literature. Arch Mal Coeur Vaiss. 1995 Jul;88(7):1021-1028. PubMed | Google Scholar
- Haj Salem N, Mesrati MA, Hadhri R, Besbes S, Belhadj M, Aissaoui A et al. Arrhythomgenic right ventricular dysplasia and sudden death: an autopsy and histological study. Ann Cardiol Angeiol (Paris). 2015;64(4):249-254. PubMed | Google Scholar
- Te Riele AS, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013 Dec;24(12):1311-20. PubMed | Google Scholar
- Saguner AM, Brunckhorst C, Duru F. Arrhythmogenic ventricular cardiomyopathy: a paradigm shift from right to biventricular disease. World J Cardiol. 2014 Apr 26;6(4):154-174. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
