
Vitiligo révélant une maladie de Vogt-Koyanagi-Harada
Mohamed El Amraoui, Youssef Zemmez, Ahmed Bouhamidi, Rachid Frikh, Naoufal Hjira, Mohammed Boui
Corresponding author: Mohamed El Amraoui, Service de Dermatologie-Vénéréologie, Hôpital Militaire d’Instruction Mohammed V, Rabat, Maroc 
Received: 15 Jan 2017 - Accepted: 27 Apr 2017 - Published: 24 Jul 2017
Domain: Dermatology,Internal medicine,Neurology (general)
Keywords: Vitiligo, Vogt-Koyanagi-Harada, auto-immunité, mélanocyte, lymphocyte T
©Mohamed El Amraoui et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mohamed El Amraoui et al. Vitiligo révélant une maladie de Vogt-Koyanagi-Harada. Pan African Medical Journal. 2017;27:220. [doi: 10.11604/pamj.2017.27.220.11656]
Available online at: https://www.panafrican-med-journal.com//content/article/27/220/full
Original article 

Vitiligo révélant une maladie de Vogt-Koyanagi-Harada
Vitiligo révélant une maladie de Vogt-Koyanagi-Harada
Vitiligo revealing Vogt -Koyanagi-Harada disease
Mohamed El Amraoui1,&, Youssef Zemmez1, Ahmed Bouhamidi1, Rachid Frikh1, Naoufal Hjira1, Mohammed Boui1
1Service de Dermatologie-Vénéréologie, Hôpital Militaire d’Instruction Mohammed V, Rabat, Maroc
&Auteur correspondant
Mohamed El Amraoui, Service de Dermatologie-Vénéréologie, Hôpital Militaire d’Instruction
Mohammed V, Rabat, Maroc
Le vitiligo est une dermatose chronique auto-immune, souvent associé ou fait découvrir d'autres pathologies auto-immunes. Son association ŕ une atteinte ophtalmologique ŕ type de pan uvéite et /ou une atteinte neurologique ŕ type de méningite et/ou de l'oreille interne ŕ type de surdité détermine la maladie ou le syndrome de Vogt-Koyanagi-Harada (VKH). Nous rapportons le cas d'une jeune femme qui consultait pour des uvéites récidivantes depuis un an, Et ce n'était qu'avec l'apparition des lésions de vitiligo que le diagnostic de maladie de VKH a été évoqué et confirmé.
English abstract
Vitiligo is a chronic auto-immune skin disease, often associated or discovers other autoimmune pathologies. His association with Ophthalmological type pan uveitis and/or neurological type of meningitis and/or inner ear type of hearing loss determines the disease or Vogt -Koyanagi-Harada syndrome (VKH). We related the case of a young woman who consulted for recurrent uveitis for a year, and it was only with the onset of vitiligo lesions that VKH disease diagnosis was discussed and confirmed.
Key words: Vitiligo, Vogt-Koyanagi- Harada, autoimmunity, melanocyte, T lymphocyte

Le vitiligo est une dermatose chronique auto-immune, souvent associé ou fait découvrir d'autres pathologies auto-immunes. Son association ŕ une atteinte ophtalmologique ŕ type de pan uvéite et /ou une atteinte neurologique ŕ type de méningite et/ou de l'oreille interne ŕ type de surdité détermine la maladie ou le syndrome de Vogt-Koyanagi-Harada (VKH). Nous rapportons le cas d'une jeune femme qui consultait pour des uvéites récidivantes depuis un an, Et ce n'était qu'avec l'apparition des lésions de vitiligo que le diagnostic de maladie de VKH a été évoqué et confirmé.

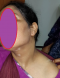
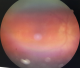
Patiente âgée de 31 ans, suivie en Ophtalmologie pour une pan uvéite évoluant par poussées et remissions depuis un an, a consulté pour des lésions de vitiligo siégeant au niveau du cou et évoluant depuis trois mois, avec une poliose du cuir chevelu sans alopécie (Figure 1). L'examen ophtalmologique montrait une baisse profonde de l'acuité visuelle des deux yeux, le tonus oculaire était normal, l'examen des annexes montrait des cils de coloration normale sans vitiligo au niveau des paupičres, l'examen du segment antérieur montrait des précipités retro dyscinétiques en graisse de mouton sans Tyndall de la chambre antérieure avec des synéchies irido-cristalliniennes et une cataracte bilatérale, le fond d'œil était trčs difficile ŕ cause d'un myosis trčs serré, les rétinographies montraient un décollement rétinien de l'œil droit et des lésions rétiniennes de l'œil gauche, une péri-vascularite témoignant d'une phlébite postérieure dans le cadre d'une uvéite postérieure (Figure 2). L'examen ORL objectivait une discrčte surdité (Figure 3). L'examen neurologique était strictement normal et les examens clinique et para clinique ŕ la recherche d'autres maladie auto-immunes associées étaient négatifs. La patiente a été mise sous corticothérapie orale et topique avec une évolution favorable.
La maladie de Harada ou syndrome de Vogt-Koaynagi-Harada, décrite par Vogt en 1906, Harada en 1926 et Koyanagi en 1929, est une pan uvéite granulomateuse bilatérale, chronique et diffuse, caractérisée par un décollement rétinien séreux et fréquemment associée ŕ des atteintes neurologiques, auditives et dermatologiques [1]. Son incidence est estimée ŕ 1/400 000 habitants avec une répartition géographique variable, au Japon elle représente prčs de 8% des uvéites. La maladie affecte essentiellement les jeunes femmes mélanodermes. Les enfants peuvent ętre affectés, mais l'âge moyen du début de la maladie est plutôt de 30 ans (de 10 ŕ 52 ans) [2,3]. La pathogenčse de l'affection a été reliée ŕ un désordre immunologique dirigé contre les mélanocytes entraînant une cytotoxicité et une apoptose médiées par les lymphocytes T. Les mélanocytes étant des cellules originaires de la cręte neurale et forment la peau, les méninges, la rétine, l'uvée, la cochlée et le labyrinthe et donc la maladie peut toucher ces différents organes. Cette maladie auto-immune qui se développerait sur un terrain génétique prédisposant est fréquemment associée avec l'haplotype DRB1*0405 [4, 5]. Cliniquement, la maladie évolue selon quatre phases: la phase prodromique: est caractérisée par des symptômes non spécifiques tels qu'une fičvre, des maux de tęte, des nausées et des vertiges, puis par des symptômes neurologiques tels qu'une faiblesse musculaire, une hémiparésie, une hémiplégie, une dysarthrie et une douleur orbitale. Durant la phase ophtalmologique: qui survient quelques jours aprčs la phase prodromique, les patients souffrent de troubles visuels, de douleur oculaire, de photophobie ou de scotome central (bilatéral dans 80 % des cas). Un décollement rétinien bilatéral et séreux survient fréquemment. Une perte de l'audition et des vertiges peuvent aussi ętre présents. La phase de convalescence qui survient dans les 3 mois aprčs le début de la maladie, est caractérisée par l'apparition de signes cutanés tels qu'une poliose affectant les cils et les sourcils (parfois les cheveux), une chute de cheveux et un vitiligo. Une uvéite récurrente et des complications ophtalmologiques apparaissent dans la derničre phase de la maladie, la phase récurrente chronique . On peut voir apparaître des néo-vaisseaux choroďdiens, un glaucome ou une cataracte secondaire [6].
Des critčres diagnostiques ont été établis et permettent de poser le diagnostic de la maladie: les critčres de Sugiura (1978) comportaient trois symptômes majeurs pour porter le diagnostic de VKH: uvéite antérieure bilatérale, décollements séreux rétiniens et pléiocytose. Les autres symptômes de la maladie (hypoacousie, vertiges, alopécie, poliose, vitiligo et dépigmentation du fond d'oeil) étaient considérés mineurs. Les critčres de « l'American Uveitis Society » (1980) ont fait l'objet d'une révision en 2001 et permettent de classer les formes de la maladie selon leur caractčre « complet », incomplet » ou « probable ». D'aprčs la littérature, il existe une concordance globale satisfaisante entre les classifications pour le diagnostic de VKH, les critčres de Sugiura sont performants pour le diagnostic des patients ŕ la phase aiguë ou lors des récurrences, Inversement, les critčres de « l'American Uveitis Society » permettent de porter plus aisément le diagnostic des formes chroniques [7]. Le diagnostic de la maladie de Vogt-Koyanagi-Harada est essentiellement clinique, les examens complémentaires contribuent au diagnostic de la maladie dans les formes incomplčtes et atypiques et ils sont de deux types, ceux explorant l'atteinte oculaire (angiographie ŕ la fluorescéine, tomographie en cohérence optique (OCT), échographie oculaire en mode B...), et ceux explorant les autres manifestations extra oculaires (ponction lombaire, IRM cérébrale, audiogramme...) [8]. Les diagnostics différentiels peuvent ętre discutés en fonction de la présentation clinique de la maladie, ainsi, Il faut différencier cette maladie d'une choriorétinopathie séreuse centrale, des métastases choroďdiennes, d'une ophtalmie sympathique, des autres causes des uvéites, du syndrome d'Alezzandrini ( hypoacousie précédant la surdité, rétinite pigmentaire, vitiligo et poliose d'un seul coté) et du syndrome Retinitis pigmentosa associated with hearing loss, thyroid disease, vitiligo, and alopecia areata. Le traitement corticoďde ŕ fortes doses est en général efficace et doit ętre prolongé pour éviter les rechutes, les immunosuppresseurs et les biothérapies peuvent également ętre utilisées. Si la prise en charge est précoce et le traitement est agressif, le pronostic est en général favorable, mais une altération sévčre de l'audition et de la vue peut survenir [9].
La maladie de Vogt-Koyanagi-Harada est une maladie auto-immune dirigée contre les mélanocytes et peut toucher alors l'uvée, les méninges, l'oreille interne et la peau. Sa prise en charge doit faire intervenir une équipe pluridisciplinaire associant ophtalmologiste, neurologue, ORL et dermatologue ŕ fin de prévenir certaines complications compromettant le pronostic fonctionnel plus que vital.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont participé activement ŕ l'élaboration de ce travail. Les auteurs déclarent avoir lu et confirmé la version finale de ce travail.
Figure 1: lésions de vitiligo avec poliose du cuir chevelu
Figure 2: rétinographie: péri-vascularite témoignant d’une phlébite postérieure dans le cadre d’une uvéite postérieure
Figure 3: audiogramme de la patiente objectivant une discrčte surdité
- Herbort Carl P, Mochizuki Manabu. Vogt-Koyanagi-Harada disease: inquiry into the genesis of a disease name in the historical context of Switzerland and Japan. Int Ophthalmol. 2007 Apr-Jun; 27(2-3):67-79. PubMed | Google Scholar
- Alaoui FZ. Syndrome de Vogt-Koyanagi-Harada ŕ propos de huit cas. La Revue de Médecine Interne. April 2007; 28(4) :Pages 250-254. Google Scholar
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada disease in children. Eye (Lond). 2008 Sep; 22(9): 1124-31. PubMed | Google Scholar
- Rao NA. Pathology of Vogt-Koyanagi-Harada disease. Int Ophthalmol. 2007 Apr-Jun;27(2-3):81-5. PubMed | Google Scholar
- Abad S, Monnet D, Caillat-Zucman S et al. Characteristics of Vogt-Koyanagi-Harada disease in a French cohort: ethnicity, systemic manifestations, and HLA genotype data. Ocul Immunol Inflamm. 2008 Jan-Feb;16(1):3-8. PubMed | Google Scholar
- Al Dousary S. Auditory and vestibular manifestations of Vogt-Koyanagi-Harada disease. J Laryngol Otol. 2011 Feb; 125 (2):138-41. PubMed | Google Scholar
- Rao NA, Sukavatcharin S, Tsai JH. Vogt-Koyanagi-Harada disease diagnostic criteria. Int Ophthalmol. 2007 Apr-Jun;27(2-3):195-9. PubMed | Google Scholar
- Zhao C, Zhang MF, Dong FT et al. Spectral domain optical coherence tomography of Vogt-Koyanagi-Harada disease: novel findings and new insights into the pathogenesis. Chin Med Sci J. 2012 Mar;27(1):29-34. PubMed | Google Scholar
- Errera MH, Fardeau C, Cohen D et al. Effect of the duration of immunomodulatory therapy on the clinical features of recurrent episodes in Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2011 Jun; 89(4):e357-66. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
PlumX Metrics
Vitiligo révélant une maladie de Vogt-Koyanagi-HaradaRecently from the PAMJ








Authors´ services
