
Le profil clinique et immunologique de 15 patients Marocains atteints de syndrome hyper IgM
Hind Ouair, Ibtihal Benhsaiene, Leila Jeddane, Jalila El Bakkouri, Naima Elhafidi, Noureddine Rada, Jilali Najib, Fatima Ailal, Hanane Salih Alj, Ahmed Aziz Bousfiha
Corresponding author: Hind Ouair, Laboratoire de Biologie et Santé, Unité de Recherche Associée au CNRST-URAC 34, Faculté des Sciences Ben M'Sik, Université Hassan II Mohammedia, Casablanca, Maroc 
Received: 17 Jun 2016 - Accepted: 03 Jan 2017 - Published: 19 Apr 2017
Domain: Clinical medicine
Keywords: Syndrome hyper IgM, clinique, immunologie, thérapie
©Hind Ouair et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hind Ouair et al. Le profil clinique et immunologique de 15 patients Marocains atteints de syndrome hyper IgM. Pan African Medical Journal. 2017;26:212. [doi: 10.11604/pamj.2017.26.212.10081]
Available online at: https://www.panafrican-med-journal.com//content/article/26/212/full
Original article 

Le profil clinique et immunologique de 15 patients Marocains atteints de syndrome hyper IgM
Le profil clinique et immunologique de 15 patients Marocains atteints de syndrome hyper IgM
Clinical and immunological profile of 15 Moroccan patients with Hyper IgM syndrome
Hind Ouair1,&, Ibtihal Benhsaien2, Leila Jeddane2, Jalila El Bakkouri2, Naima Elhafidi3, Noureddine Rada4, Jilali Najib5, Fatima Ailal2, Hanane Salih Alj1, Ahmed Aziz Bousfiha2
1Laboratoire de Biologie et Santé, Unité de recherche Associée au CNRST-URAC 34, Faculté des Sciences Ben M'Sik, Université Hassan II Mohammedia, Casablanca, Maroc, 2Laboratoire d'Immunologie Clinique, Inflammation et Allergie, Faculté de Médecine et Pharmacie, Université Hassan II, Casablanca, Maroc, 3Département de Pédiatrie 1, Hôpital d'Enfants de Rabat, CHU Ibn Sina, Rabat, Maroc, 4Département de Pédiatrie, CHU Mohamed VI, Marrakech, Maroc, 5Département des Maladies Infectieuses, Hôpital A. Harouchi, CHU Ibn Rochd, Casablanca, Maroc
&Auteur correspondant
Hind Ouair, Laboratoire de Biologie et Santé, Unité de Recherche Associée au
CNRST-URAC 34, Faculté des Sciences Ben M'Sik, Université Hassan II Mohammedia,
Casablanca, Maroc
Le Syndrome hyper IgM est un déficit immunitaire héréditaire bien connu, décrit pour la premičre fois en 1961. Il est causé par un défaut au niveau des lymphocytes B, caractérisé par un taux sérique normal ou élevé des IgM et un taux bas ou nul des IgG, IgA, IgE résultant d'une déficience de la commutation isotypique. Ses manifestations cliniques sont dominées par les infections ŕ répétition, surtout du tube digestif, de la sphčre ORL et des poumons. Le syndrome est causé par un défaut de commutation de classe d'immunoglobuline dans les cellules B, et une diminution de la capacité des monocytes ŕ induire la prolifération des lymphocytes T. Le résultat net de tous ces défauts est la susceptibilité accrue aux infections opportunistes ŕ Pneumocystis jiroveci, Cryptosporidium spp et d'autres organismes intracellulaires, ainsi qu'une fréquence élevée d'infections bactériennes et virales. L'intéręt de ce projet est d'illustrer l'importance de la compréhension des mécanismes physiopathologiques associés ŕ cette susceptibilité accrue aux infections, ce qui permettra une meilleure prise en charge diagnostique et thérapeutique des patients atteint du Syndrome hyper IgM (SHIM).
English abstract
Hyper IgM syndrome is a well known genetic (primary) immunodeficiency disorder which was first described in 1961. It is caused by B lymphocyte deficiency characterized by normal or elevated serum IgM levels and low or zero levels of IgG, IgA, IgE resulting from isotype-switching deficiency. Clinical manifestations are dominated by recurrent infections, especially involving the digestive tube of the ENT sphere and the lungs. This syndrome is caused by B-cell immunoglobulin class switch deficiency and decreased capacity to induce proliferation of T lymphocytes. The net result of these deficiencies is reflected in increased susceptibility to Pneumocystis jiroveci, Cryptosporidium spp and other intracellular organisms as well as high rate of bacterial and viral infections. This study aimed to illustrate the importance of understanding the pathophysiological mechanisms associated with this increased susceptibility to infections in order to allow a better diagnosis and therapy in patients with Hyper IgM syndrome (HIM).
Key words: Hyper IgM syndrome, clinic, immunology, therapy

Le syndrome hyper IgM (SHIM) ou Class Switch Recombi¬nation Deficiencies (CSR-Ds) constitue un groupe de maladies génétiquement hétérogčne. Il est causé par un défaut au niveau de la commutation isotypique en association ou non avec un défaut d'hypermutation somatique dans les lymphocytes B [1]. Les CSR-D sont classés en 6 types, en fonction du gčne et de la protéine mutée: SHIM1 (CD40L), SHIM2 (AID), SHIM3 (CD40), SHIM5 (Ung), SHIM6 (NEMO) et SHIM7 (IKBA) [2]. Les manifestations cliniques se développent pendant la premičre ou la deuxičme année de vie pour la majorité des patients et elles sont dominées essentiellement par des infections récurrentes (pulmonaires, ORL, digestives), des infections opportunistes, des signes d'auto-immunité et de lymphoproliferation. Le bilan immunitaire montre un taux bas des IgG, A et E. Une neutropénie d'origine autoimmune peut également ętre observée.

Notre étude porte sur 15 cas de CSR-D colligés ŕ l'Unité d'Immunologie Clinique du service de Pédiatrie 1 de l'Hôpital d'Enfants A. Harouchi, CHU Ibn Rochd de Casablanca, depuis Novembre 1995 jusqu'ŕ Décembre 2015. Cette étude a inclus tous les enfants présentant un CSR-D dont l'âge au diagnostic varie de 7 jours ŕ 15 ans. Le diagnostic était suspecté devant une histoire clinique évocatrice de déficit immunitaire, et était confirmé par un bilan immunitaire, comportant une sérologie VIH, le typage des sous populations lymphocytaires (SPL) et le dosage pondéral des Immunoglobulines A, G et M par néphélométrie.
Notre étude a pu colliger 15 patients (7 filles et 8 garçons) atteints de CSR-D (Défaut de Commutation Isotypique) issus de 13 familles différentes, dont 12 cas sont issus d'un mariage consanguin. L'âge d'apparition des premiers signes varie entre 7 jours et 16 mois. L'âge de diagnostic est compris entre 5 mois et 12 ans avec un âge médian de 36 mois. Des infections respiratoires ŕ répétition sont le signe révélateur chez 15 patients, soit 88,8%, compliquées de dilatation de bronches chez 6 cas et de pneumatocčle dans un cas. Le tableau clinique était complété par des infections de la sphčre ORL (66,6% des cas), des infections digestives entrainant des diarrhées chroniques (27,7%), un Syndromelymphoprolifératif (11 patients) dont hépatomégalie chez 5 cas, splénomégalie chez 5 cas et adénopathies périphériques dans 6 cas. L'atteinte cutanée est notée chez 5 cas de type abcčs de la face, des verrues multiples chez 2 cas, des lésions cutanées érythémato-papuleuses généralisées récidivantes dans 1 cas (Tableau 1). Tous les malades ont eu une sérologie VIH négative. Les SPL sont normales dans tous les cas. Quinze enfants de notre série avaient un taux élevé d'IgM ŕ une moyenne de 3,81 g/L contre 2 malades avec un taux normal. Sur 15 patients marocains cliniquement diagnostiqués comme HIGM, 8 sont encore vivants, sous thérapie de remplacement réguličre, associé ŕ une antibioprophylaxie adéquate (Tableau 2).
Les syndromes hyper-IgM sont des déficits immunitaires extręmement rares, décrit pour la premičre fois en 1961 par Rosen et al d'une façon indépendante [3]. Plusieurs causes moléculaires sont aujourd'hui connues pour expliquer et classer les syndromes HIGM qui peuvent ętre soit un défaut de signalisation entre les lymphocytes T et B, soit de défauts de transmission intracellulaire du signal induit par l'interaction entre le CD40L et le CD40. Le syndrome hyper IgM lié ŕ l'X représenterait 65 ŕ 70% des syndromes hyper-IgM [4], c'est le plus fréquent en Europe oů le taux de consanguinité est trčs bas et c'est le plus étudié. Le registre américain rapporte que le taux d'incidence minimale de l'X-HIGM était d'environ 1 cas pour 1.030.000 naissances vivantes [5]. Le déficit en AID vient en 2čme position de fréquence aprčs l'X-HIGM. Minegishi a suggéré que 50% des syndromes hyper IgM non lié ŕ l'X étaient des syndromes HIGM2 [6]. Les autres formes, autosomique récessive ou dominante, constituent un groupe hétérogčne sur le plan clinique. Aux Etats Unis (2005), dans une large cohorte américaine de 140 patients (dont 130 garçons) ayant un syndrome HIGM, Lee et al rapportent un déficit en CD40L chez 98 des cas (70%), un déficit en AID chez 4 malades (3%), et un défaut du signal NF?B chez un autre malade. Aucune mutation n'a pu ętre identifiée chez 33 patients (24%) [7]. En Asie, dans une cohorte Iranienne de 33 malades (dont 28 garçons et 5 filles), la consanguinité était notée parmi 9 malades et le profil moléculaire était en faveur des formes autosomiques récessives étant donné le taux important de consanguinité et le phénotype clinique [8]. Alors que dans une série Taďwanaise, Lee (2014) rapporte 14 patients avec un syndrome HIGM appartenant ŕ 12 familles différents dont 10 cas sont identifiés comme formes liées ŕ l'X [9]. Au Maghreb, une étude tunisienne rapportée par Mellouli en 2012 sur 710 cas de DIP colligés entre Avril 1988 et Avril 2012 a montré que les syndromes Hyper IgM représentaient 4,2% des DIP en Tunisie [10]. Sur 502 cas de DIP enregistrés, au Maroc, jusqu'ŕ 2013, les syndromes HIGM représentaient 2.8% des DIP [11]. Une autre étude sur 51 DIP colligés au CHU de Sfax entre 1995 et 2010 a montré que les syndromes Hyper-IgM représentaient 9.8 % de l'ensemble des DIP diagnostiqués [12]. Les patients inclus dans cette étude ont présenté des infections récidivantes caractérisées par leur précocité et gravité. Il s'agit d'infections respiratoires chez 58,8% des cas, dominées par des pneumopathies; des infections du tractus gastro-intestinales révélées par des diarrhées chroniques chez 5 cas (30%); des infections ORL, otites moyennes aigues (30%) et candidose buccale (23,5%).
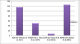
Ainsi, l'âge de diagnostic et les manifestations clinique varient selon le type du SHIM (Tableau 3, Figure 1) [1,13] surviennent au bas âge dčs la premičre année de vie voire dans les premiers mois de vie avec un âge médian de 12 mois [14]. Les patients avec AR-HIGM sont plus âgés au moment du diagnostic que ceux ayant un syndrome HIGM1 ou un syndrome HIGM3. Ainsi pour le syndrome HIGM 2, l'âge est compris entre 1 et 13 ans pour les patients de Revy [13] et entre 2 et 28 ans pour ceux de Minegishi [6]. Dans notre série l'âge médian de diagnostic était de 36 mois. Dans le Registre européen XHIGM, les adénopathies généralisées ont été rapportées dans 7 cas sur 56 patients [15]. L'évolution dans le syndrome HIGM1 est marquée par la répétition des pneumopathies et l'installation de bronchectasies. Un retard de croissance a été rapporté chez certains patients, il est dű essentiellement aux infections ŕ répétition et au retard diagnostique [15]. De plus, les patients avec X-HIGM sont plus fréquemment sujets ŕ développer des Arthrites séronégatives, une encéphalopathie dégénérative, une hypothyroďdie, une néphropathie auto-immune. Les hyperplasies lymphoďdes sont fréquentes dans le syndrome HIGM2: 13 cas sur 18 dans l'étude de Revy, 9 cas sur 18 dans l'étude de Minegishi et les manifestations auto-immunes, (diabčte, polyarthrite, hépatite auto-immune, anémie auto-immune, thrombocytopénie, maladie de Crohn, et uvéite chronique) sont observées chez 20 cas sur 29 dans la série de Quartier (Tableau 4) [1, 2, 16, 17]. En dehors des gčnes décrits ci-dessus, d'autres défauts moléculaires de la commutation isotypique pouvant conduire ŕ un phénotype HIGM ont été identifiés chez des patients ayant un autre type de DIP avec des mutations des enzymes impliquées dans la réparation de l'ADN: la protéine PMS2, la protéine ATM impliquée dans l'ataxie-télangiectasie [18]. La thérapie de remplacement d'Ig constitue le traitement principal pour corriger les conséquences cliniques du déficit humoralprésent dans toutes les formes du syndrome HIGM. Dans les syndromes HIGM 2, HIGM 4 et HIGM 5, le traitement est basé essentiellement sur les Ig IV, qui réduit sensiblement l'incidence des infections bactériennes. Dans les syndromes HIGM1 et HIGM3, le traitement par les IgIV et l'antibioprophylaxie ŕ base TSU est indiqué chez la majorité des patients en attente de la greffe de CSH, ce qui permet de réduire la fréquence et la gravité des infections mais ne prévient pas la maladie lymphoproliférative, la cholangite sclérosante et les affections malignes [19].
En conclusion, cette étude a pu rassembler tous les patients HIGM diagnostiqués au sein de l'Unité d'Immunologie clinique, ils présentent des signes cliniques diverses dominés par des Infections respiratoires et des taux normaux ou élevé des IgM.
Etat des connaissances actuelles sur le sujet
- Le syndrome hyper IgM (SHIM) est une maladie génétiquement hétérogčne;
- Il s'agit d'un défaut de la commutation isotypique;
- Se manifeste par des infections récurrentes (pulmonaires, ORL, digestives).
Contribution de notre étude à la connaissance
- Caractéristiques cliniques des patients marocains pris en charge ŕ l'Hôpital d'enfants AbderahimHarouchi-Casablanca (Centre de Référence des Déficits Immunitaires Primitifs);
- Effet de la consanguinité sur l'évolution de ce syndrome;
- Le suivi et l'évolution de ces patients atteints de syndrome Hyper IgM.
Les auteurs ne déclarent aucun conflit d'intéręts.
Hind OUAIR: revue bibliographique, analyse de données, interprétation des résultats et rédaction du manuscrit. Ibtihal BENHSEINE: examen clinique, interprétation des résultats, correction du manuscrit. Leila JEDDANE: interprétation des résultats et correction du manuscrit. Jalila EL BAKKOURI: phénotypage des sous populations lymphocytaires. Naima ELHAFIDI: examen clinique. Noureddine RADA: examen clinique. Jilal NAJIB, Fatima AILAL: conception, examen clinique, analyse des données et interprétation des résultats. Hanane SALIH ALJ: conception analyse des données, interprétation des résultats et correction du manuscrit. Ahmed Aziz BOUSFIHA: conception, examen clinique, analyse des données, interprétation des résultats et correction du manuscrit du manuscrit. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Ils vont ŕ l'endroit de Ridha BARBOUCH, Bandar AL SAOUD et BEJAOUI Med pour le contribution.
Tableau 1: description cliniques des patients marocains atteints de Syndrome Hyper IgM
Tableau 2: bilan immunologique
Tableau 3: la démographie et description clinique des déficits en CD40
Tableau 4: âge de diagnostic, signes révélateurs et microorganismes dans différentes cohortes du SHIM
Figure 1: âge médian de diagnostic selon le type des CSR-D
- Tang WJ, An YF, Dai RX, Wang QH, Jiang LP, Tang XM, Yang XQ, Yu J, Tu WW, Zhao XD. Clinical, molecular, and T cellsubset analyses in a smallcohort of Chinese patients with hyper-IgM syndrome type Hum Immunol. 2014; 75(7):633-40. PubMed | Google Scholar
- Madkaikar M, Gupta M, Chavan S, Italia K, Desai M, Merchant R, Radhakrishnan N, Ghosh K. X-linked hyper IgM syndrome: clinical, immunological and molecularfeatures in patients fromIndia. Blood Cells Mol Dis. 2014; 53(3):99-104. PubMed | Google Scholar
- Rosen FS, Kevy SV, Merler E, Janeway CA, Gitlin D.Recurrentbacterial infections and dysgamma-globulinemia: deficiency of 7S gamma-globulins in the presence of elevated 19S gamma-globulins: report of two cases. Pediatrics. 1961; 28:182-95. PubMed | Google Scholar
- Etzioni A, Ochs HD. The hyper IgM syndrome--an evolving story. PediatrRes. 2004; 56(4):519-25. PubMed | Google Scholar
- Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, Conley ME. The X-linked hyper-IgM syndrome: clinical and immunologicfeatures of 79 patients. Medicine. 2003; 82(6):373-84. PubMed | Google Scholar
- Minegishi Y, Lavoie A, Cunningham-Rundles C, Bédard PM, Hébert J, Côté L, Dan K, Sedlak D, Buckley RH, Fischer A, Durandy A, Conley ME. Mutations in activation-inducedcytidine deaminase in patients with hyper IgM syndrome. Clin Immunol. 2000;97(3):203-10. PubMed | Google Scholar
- Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005 ;105(5):1881-90. PubMed | Google Scholar
- Abolhassani H, Akbari F, Mirminachi B, Bazregari S, Hedayat E, Rezaei N, AghamohammadiA.Morbidity and mortality of Iranian patients with hyper IgM syndrome: a clinical analysis. Iran J Immunol. 2014 ; 11(2):123-33. PubMed | Google Scholar
- Lee WI, Huang JL, Yeh KW, Yang MJ, Lai MC, Chen LC, Ou LS, Yao TC, Lin SJ, Jaing TH, Chen SH, Hsieh MY, Yu HH, Chien YH, Shyur SD. Clinical features and genetic analysis of Taiwanese patients with the hyper IgM syndrome phenotype. Pediatr Infect Dis J. 2013 ;32(9):1010-6. PubMed | Google Scholar
- Mellouli F, Mustapha IB, Khaled MB, Besbes H, Ouederni M, Mekki N, Ali MB, Largučche B, Hachicha M, Sfar T, GueddicheN,Barsaoui S, Sammoud A, Boussetta K, Becher SB, Meherzi A, Guandoura N, Boughammoura L, Harbi A, Amri F, Bayoudh F, Jaballah NB, Tebib N, Bouaziz A, Mahfoudh A, Aloulou H, Mansour LB, Chabchoub I, Boussoffara R, Chemli J, Bouguila J, Hassayoun S, Hammami S, Habboul Z, Hamzaoui A, Ammar J, Barbouche MR, Bejaoui M. Report of the Tunisian Registry of Primary Immunodeficiencies: 25 Years of Experience (1988-2012). J Clin Immunol. 2015; 35(8):745-53. PubMed | Google Scholar
- Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, Kili A, Benmiloud S, Benhsaien I, Faiz I, Maataoui O, Aadam Z, Aglaguel A, Baba LA, Jouhadi Z, Abilkassem R, Bouskraoui M, Hida M, Najib J, Alj HS. First report on the Moroccanregistry of primaryimmunodeficiencies: 15 years of experience (1998-2012). J Clin Immunol. 2014 May;34(4):459-68. PubMed | Google Scholar
- Lamia S, Aloulou H, Kamoun T, Chabchoub I, Ben Moustapha I, Barbouch R, Mongia H. [Primaryimmunodeficiencydisorders in 51 cases]. Tunis Med. 2013; 91(1):38-43. PubMed | Google Scholar
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 2000; 102(5):565-575. PubMed | Google Scholar
- Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, Resnick I, Fasth A, Baer M, Gomez L, Sanders EA, Tabone MD, Plantaz D, EtzioniA,Monafo V, Abinun M, Hammarstrom L, Abrahamsen T, Jones A, Finn A, Klemola T, DeVries E, Sanal O, Peitsch MC, Notarangelo LD. Clinicalspectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997; 131(1 Pt 1):47-54. PubMed | Google Scholar
- Karaca NE, Forveille M, Aksu G, Durandy A, Kutukculer N. Hyper-immunoglobulin M syndrome type 3 with normal CD40 cell surface expression. Scand J Immunol. 2012; 76(1):21-5. PubMed | Google Scholar
- Durandy A, Revy P, Imai K, Fischer A. Hyper immunoglobulin M syndromes caused by intrinsic B-lymphocyte defects. ImmunolRev. 2005; 203:67-79. PubMed | Google Scholar
- Lopez-Granados E, Temmerman ST, Wu L, Reynolds JC, Follmann D, Liu S, Nelson DL, Rauch F, Jain A. Osteopenia in X-linked hyper-IgM syndrome reveals a regulatory role for CD40 ligand in osteoclastogenesis. Proc NatlAcadSci U S A. 2007; 104(12):5056-61. PubMed | Google Scholar
- Quartier P, Bustamante J, Sanal O, Plebani A, Debré M, Deville A, Litzman J, Levy J, Fermand JP, Lane P, Horneff G, Aksu G, Yalçin I, Davies G, TezcanI,Ersoy F, Catalan N, Imai K, Fischer A, Durandy A. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clin Immunol. 2004; 110(1):22-9. PubMed | Google Scholar
- Durandy A, Kracker S. The Hyper IgM Syndromes–a Long List of Genes and Years of Discovery. Elsevier AcademicPress. 2014;198-210. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
