
Fibrodysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A mutation
Mohammed Saleh, Joost Commandeur, Renata Bocciardi, Grace Kinabo, Ben Hamel
Corresponding author: Ben Hamel, Department of Human Genetics, Radboud university medical center, P.O. Box 9101, 6500 HB Nijmegen, The Netherlands 
Received: 24 Sep 2015 - Accepted: 14 Nov 2015 - Published: 24 Nov 2015
Domain: Clinical medicine
Keywords: Fibrodysplasia ossificans progressiva, heterotopic ossification, hallux valgus, recurrent ACVR1 mutation
©Mohammed Saleh et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mohammed Saleh et al. Fibrodysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A mutation. Pan African Medical Journal. 2015;22:299. [doi: 10.11604/pamj.2015.22.299.8032]
Available online at: https://www.panafrican-med-journal.com//content/article/22/299/full
Original article 

Fibrodysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A mutation
Fibrodysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A mutation
Mohammed Saleh1, Joost Commandeur1,2, Renata Bocciardi3,4, Grace Kinabo1, Ben Hamel5,&
1Department of Paediatrics and Child Health, Kilimanjaro Christian Medical Centre, P.O. Box 2240, Moshi, Tanzania, 2Department of Internal Medicine, Kilimanjaro Christian Medical Centre, P.O. Box 2240, Moshi, Tanzania, 3Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health and CEBR, Universit� Degli Studi di Genova, Via G. Gaslini, 5, 16147 Genova, Italy, 4Istituto Giannina Gaslini, Medical Genetics Unit, Via G. Gaslini, 5, 16147 Genova, Italy, 5Department of Human Genetics, Radboud university medical center, P.O. Box 9101, 6500 HB Nijmegen, The Netherlands
&Corresponding author
Ben Hamel, Department of Human Genetics, Radboud university medical center, P.O. Box 9101, 6500 HB Nijmegen, The Netherlands
Fibrodysplasia ossificans progressiva is a rare autosomal dominantly inherited disorder of connective tissue caused by mutations in the gene encoding for ACVR1/ALK2, a bone morphogenetic protein type I receptor. It is mainly characterized by congenital malformations of the great toes and the formation of qualitatively normal bone in extra-skeletal sites leading to severe disability and eventually death. We present a sporadic case from Northern Tanzania with a minor unilateral hallux anomaly and the common ACVR1 c.617G>A mutation.
Fibrodysplasia ossificans progressiva (FOP, OMIM #135100) is a rare and disabling autosomal dominantly inherited disorder of connective tissue leading to progressive development of heterotopic ossification (HO) in extra-skeletal sites, i.e. the formation of qualitatively normal bone in skeletal muscles and other connective tissues [1]. We present a case of FOP that was diagnosed clinically, though only a minor unilateral hallux anomaly was present, in the paediatric department of Kilimanjaro Christian Medical Centre (KCMC) in Moshi, Tanzania, almost 50 years after the first FOP case was reported from Tanzania [2]. The case we present here is of interest because of the minimal and unilateral hallux abnormality in the presence of the common ACVR1 c.617G>A mutation. Also, it is the first molecularly confirmed case of FOP in sub-Saharan Africa outside South Africa.
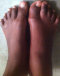
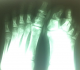
In October 2013, a 12-year-old girl was referred to the paediatric clinic of KCMC with fever, weakness and headache for seven days, which was diagnosed as malaria and treated accordingly. She had a history of restricted movements of arms and neck. She had pain and swelling involving the neck, shoulders and back areas, which began when she was two years of age. Over the following ten years she had repeated episodes of swelling involving the neck, shoulder and back coupled with increasing difficulty walking. At ten years of age she also started to experience difficulty with eating and talking because of decreased motility of the jaw. No history of trauma was reported. She is the seventh child in her family of eight children and the only affected in her extended family. On examination the patient was febrile with distinctive hard non-tender swellings involving the neck, shoulder and the upper part of the back (Figure 1). These abnormalities were associated with limited range of movements over the jaw, neck, shoulders and elbow joints. In addition the patient was unable to extend her legs because of contractures of the hamstring tendons. Thumbs were normal. A slight hallux valgus of the right great toe was noted (Figure 2) and confirmed radiologically (Figure 3). Additional X-rays confirmed the presence of HO lesions around the shoulder and pelvic girdles, elbows and back. A clinical diagnosis of FOP was made and a venous blood sample was obtained. Molecular analysis of the ACVR1/ALK2 gene revealed the presence of the most frequently recurrent c.617G>A (p.Arg206His) mutation, confirming the clinical diagnosis. She was given paracetamol for the pain and continued to be seen in our clinic.
FOP is a rare disabling inherited disorder of connective tissue with a prevalence of 0,5 per 1 million without apparent racial, ethnic or geographical variation [1]. It is most frequently caused by a recurrent heterozygous gain-of-function mutation (c.617G>A; p.Arg206His) in the ACVR1/ALK2 gene on chromosome 2q23, which usually occurs as a sporadic, de novo mutation, but it may be inherited from either parent [3,4]. Other mutations may lead to a variant phenotype [5]. Mutations cause an enhanced BMP-mediated signalling leading to progressive development of HO in skeletal muscles and other connective tissues, i.e. the formation of qualitatively normal bone in extra-skeletal sites [1]. The case reported in this study appears to be sporadic, since neither parent nor other first-degree relatives are known to be affected. When present, FOP can be diagnosed clinically early in life and even prenatally [6], by the presence of short malformed halluces (monophalangism, hallux valgus and/or malformed first metatarsal). Sometimes similar malformations of the thumbs are also present. Our case had just a minor and unilateral hallux valgus and still the common ACVR1 c.617G>A mutation. Though this has been reported before, it is rare [1]. The disease progresses with sporadic exacerbations (flare-ups) resulting in HO, which starts in the dorsal, axial, cranial and proximal regions of the body. The heart, smooth muscles and the diaphragm are most notably not affected by this disease. Flare-ups can be induced by different triggers such as trauma, surgery, diagnostic biopsies, intramuscular injections and viral infections. Also, general anaesthesia poses dangers to patients with FOP [7]. FOP is often misdiagnosed [8]. From Nigeria a case was reported where FOP was misdiagnosed as Burkitt�s lymphoma [9]. We know of another case in our hospital which was misdiagnosed as Burkitt's lymphoma and in whom a diagnostic liver biopsy contributed in our opinion to the development of HO at the abdominal wall. Another article from Nigeria demonstrated the negative effect of surgical intervention in children with undiagnosed FOP [10]. These articles show the iatrogenic harm that can be done by not recognising FOP in time. HO eventually leads to impaired mobility, weight loss due to ankylosis of the jaw and thoracic insufficiency due to costovertebral malformations. Median age of survival is 40 years [11], however, we hypothesize that this may be lower in sub-Saharan African patients due to delayed diagnosis and higher risks of trauma and infections. Currently, there is no effective prevention or cure for this disabling disease.
Early diagnosis of FOP, which in cases with a minor and unilateral hallux anomaly supposes a high level of awareness, is important in order to prevent flare-ups, for example by restricting activities to reduce the risk of trauma and by reducing the number of unnecessary (invasive) investigations and interventions, thereby preventing iatrogenic harm [1,8,10].
The authors declare no competing interests.
MS and GK diagnosed and managed the patient. MS helped draft the manuscript. BH and JC conceived the article and wrote the manuscript. RB carried out the molecular genetic studies. All authors read and approved the final manuscript.
We are thankful for the contributions of Drs Marieke Dekker and William Howlett of Kilimanjaro Christian Medical Centre, Moshi, Tanzania in preparing this article. Prof Roberto Ravazzolo of Istituto Giannina Gaslini, Genova, Italy is gratefully acknowledged for allowing the molecular analysis to be performed in his laboratory free of charge.
Figure 1: FOP lesions on the back
Figure 2: hallux valgus
of the right great toe
Figure 3: radiograph, showing the hallux valgus of the right great toe
- H�ning I, Gillessen-Kaesbach G. Fibrodysplasia ossificans progressiva: clinical course, genetic mutations and genotype-phenotype correlation. Mol Syndromol. 2014;5(5):201-1. PubMed | Google Scholar
- Ebrahim GJ, Grech P, Slavin G. Myositis ossificans progressiva in an African child. Br J Radiol.1966;39(468):952-3. PubMed | Google Scholar
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressive. Nat Genet. 2006;38(5):525-7. PubMed | Google Scholar
- Bocciardi R, Bordo D, Di Duca M, Di Rocco M, Ravazzolo R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet. 2009;17(3):311-8. PubMed | Google Scholar
- Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG, Brown MA, Wordsworth BP, Triffitt JT. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS One. 2009;4(3):e500. PubMed | Google Scholar
- Maftei C, Rypens F, Thiffault I, Dub� J, Laberge AM, Lemyre E. Fibrodysplasia ossificans progressiva: bilateral hallux valgus on ultrasound a clue for the first prenatal diagnosis for this condition-clinical report and review of the literature. Prenat Diagn. 2015;35(3):305-7.. PubMed | Google Scholar
- Liu JX, Hu R, Sun Y, Jiang H. General anesthesia in fibrodysplasia ossificans progressive: a case report and clinical review. Int J Clin Exp Med. 2014;7(5):1474-9. PubMed | Google Scholar
- Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressive. Pediatrics. 2005;116(5):e654-61. PubMed | Google Scholar
- Ejeckam GC, Ibekwe O. Myositis ossificans masquerading as Burkitt's lymphoma of maxilla in a Nigerian boy.Trop Geogr Med.1981;33(2):197-9. PubMed | Google Scholar
- Obamuyide HA, Ogunlade SO. A Tumour for which Surgery will do more harm than good: A Case Report of Fibrodysplasia Ossificans Progressiva. Niger Postgrad Med J. 2015;22(1):83-8.. PubMed | Google Scholar
- Kaplan FS, Zasloff MA, Kitterman JA, Shore EM, Hong CC, Rocke DM. Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressive. J Bone Joint Surg Am. 2010;92(3):686-91. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ




Authors´ services
