
Une maladie immunoproliférative de l’intestin gręle révélée par une invagination intestinale aigue: ŕ propos d’un cas
Amal Bennani, Kaoutar Znati, Salima Rezzouk, Hicham Bouhadouti, Khalid Maazaz, Affaf Amarti
Corresponding author: Bennani Amal, Service d’Anatomie Pathologique, CHU Hassan II, Fčs, Maroc 
Received: 27 Oct 2013 - Accepted: 03 Dec 2013 - Published: 14 Jan 2014
Domain: Clinical medicine
Keywords: Maladie immunoproliférative, intestin grêle, invagination intestinale
©Amal Bennani et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Amal Bennani et al. Une maladie immunoproliférative de l’intestin gręle révélée par une invagination intestinale aigue: ŕ propos d’un cas. Pan African Medical Journal. 2014;17:12. [doi: 10.11604/pamj.2014.17.12.3512]
Available online at: https://www.panafrican-med-journal.com//content/article/17/12/full
Original article 

Une maladie immunoproliférative de l’intestin gręle révélée par une invagination intestinale aigue: ŕ propos d’un cas
Une maladie immunoproliférative de l’intestin gręle révélée par une invagination intestinale aigue: ŕ propos d’un cas
Amal.Bennani1,&, Kaoutar Znati1, Salima Rezzouk2, Hicham Bouhadouti2, Khalid Maazaz2, Affaf amarti1
1Service d’Anatomie Pathologique, CHU Hassan II, Fčs, Maroc 2Service de Chirurgie Viscérale, CHU Hassan II, Fčs, Maroc
&Auteur correspondant
Bennani Amal, Service d’Anatomie Pathologique, CHU Hassan II, Fčs, Maroc
La maladie immunoproliférative de l'intestin gręle IPSID est un lymphome rare qui se développé ŕ partir du systčme lymphoďde associé aux muqueuses au niveau de l'intestin gręle. Sa révélation par une invagination intestinale aigue est exceptionnelle et n'a jamais été rapporté dans la littérature auparavant. Nous rapportons le cas d'un patient de 65ans, admis aux urgences dans un tableau d'invagination intestinale aigue. Le scanner abdominal a mis en évidence masse gręlique ŕ paroi concentrique avec incarcération du segment mésentérique au sein de la lésion fortement évocatrice d'une invagination intestinale. Le patient a été opéré et a bénéficié d'une résection iléale emportant le boudin d'invagination. L'examen histologique de la masse a été en faveur d'une maladie des chaines alpha transformée en un lymphome B diffus ŕ grandes cellules. Les auteurs rapportent un cas rare d'une IPSID révélée par une invagination intestinale aigue et ŕ travers cette observation, mettent en relief les principaux aspects cliniques, histologiques, thérapeutiques de cette entité avec une revue de la littérature.
La maladie immunoproliférative de l'intestin gręle (IPSID) également appelé maladie des chaînes lourdes alpha est un lymphome qui touche le systčme des immunoglobulines A exocrine de la muqueuse et préférentiellement celui de l'intestin gręle et s'accompagne de la sécrétion d'une chaine lourde alpha. Sa révélation par une invagination intestinale aigue est exceptionnelle et n'a jamais été rapporté dans la littérature.
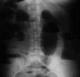
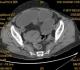
Il s'agit d'un patient âgé de 65 ans, admis au service des urgences dans un tableau d'occlusion intestinale aigue. Le patient rapporte la notion de douleurs abdominales similaires depuis un mois. L'examen clinique a révélé une distension abdominale avec une matité ŕ la percussion, et au toucher rectale une ampoule rectale vide. Une radiographie de l'abdomen sans préparation a mis en évidence de multiples niveaux hydro-aériques gręliques (Figure 1). L'échographie abdominale a mis en évidence une masse digestive pelvienne hétérogčne mesurant environ 8 cm de diamčtre. Un scanner abdominal complémentaire a montré une masse gręlique ŕ paroi concentrique avec incarcération du segment mésentérique au sein de la lésion fortement évocatrice d'une invagination intestinale (Figure 2).Le patient a été opéré et a bénéficié d'une résection iléale emportant le boudin d'invagination avec une anastomose termino-terminale. L'examen macroscopique de la pičce opératoire a mis en évidence une masse de 7 cm de diamčtre exoluminale arrondie, homogčne et friable ŕ la coupe qui s'invagine ŕ l'intérieur d'un segment iléal. L'examen histologique a retrouvé prolifération lymphomateuse faite de petites cellules avec différenciation plasmocytaire nette, intéressant toutes les couches de la paroi gręlique et arrivant jusqu'au niveau de la séreuse. Par endroit on notait la présence de cellules lymphoďdes de grande taille immunoblastiques et centroblastiques formant des nappes diffuses et parfois des clusters de 5 ŕ 10 cellules (Figure 3). Une étude immunohistochimique a été réalisée et a montré une forte expression du CD20 par les cellules tumorales (Figure 4). Devant cet aspect morphologique et immunohistochimique le diagnostic retenu est celui d'une maladie des chaines alpha transformée en un lymphome B diffus ŕ grandes cellules. Un bilan d'extension réalisé s'est révélé négatif.
Les lymphomes digestifs constituent la premičre localisation extra-ganglionnaire des lymphomes et sont, dans l´immense majorité des cas, de type non hodgkinien [1]. Parmi ces lymphomes on ditingue les IPSID (immuno-proliferative small intestinal diseases) qui sont des proliférations diffuses du systčme lymphoďde B du tube digestif caractérisés par une atteinte diffuse du gręle sans intervalle de muqueuse saine [2,3].
Ce lymphome affecte les jeunes des deux sexes entre 15 et 30 ans mais il peut atteindre les enfants et les personnes âgées [4]. La majorité des cas recensés provient du bassin méditerranéen et du moyen orient mais de nombreux cas ont été observés chez les malades originaires de l'Europe de l'est, et de l'ex URSS et de l'Amérique centrale et du sud [4]. Le seul dénominateur commun de tous ces malades étant la mauvaise hygične et le niveau socio-économique bas comme c'est le cas pour notre patient. Lecuit et al ont prouvé grâce ŕ la technique de PCR l'association entre le Campylobacter jéjuni et l'IPSID [5]. La toxine cytoléthale du C. jejuni engendre des cassures double-brin de l'ADN qui favorisent le développement de cellules B mutées, différenciées en cellules plasmocytaires aberrantes produisant des chaînes lourdes tronquées. L'absence de région variable ŕ la surface des cellules B mutées explique qu'elles échappent au systčme immunitaire, avec pour conséquence une prolifération responsable du développement d'IPSID [6].
L'IPSID se manifeste habituellement par une malabsorption associée ŕ une entéropathie exsudative [7]. Elle peut se manifester également ŕ l'occasion de douleurs abdominales, ou de vomissements. A un stade tardif, la maladie peu prendre une allure tumorale avec une véritable masse abdominale responsable d'une urgence chirurgicale telle que l'invagination intestinale aigue comme c'est le cas de notre patient.
Sur le plan biologique, l'immunofixation des protides sanguins révčle une protéine sérique anormale dans 40 ŕ 100 % des cas, qui correspond ŕ des chaînes lourdes alpha sériques. Sa détection est plus importante au stade des IPSID de bas grade, plus rare au niveau des cellules immunoblastiques (stade plus avancé de la maladie).
Un bilan endoscopique doit ętre réalisé pour mettre en évidence l'étendue des lésions et pour obtenir le diagnostic histologique [8,9].
La vidéocapsule endoscopique [10] est une technique non invasive et performante, grâce ŕ laquelle l'intestin gręle est le segment du tube digestif le mieux étudié avec une sensibilité meilleure et une rentabilité diagnostique de 50-70 %. Des biopsies multiples, étagées doivent ętre réalisées : fixées dans du formol pour une étude immunohistochimique et de biologie moléculaire, la congélation peut ętre recommandée (dans le cadre d'essais cliniques).
La stadification utilisée est celle d'Ann Arbor, modifiée par Musshoff pour le tube digestif. Dans la classification de l'OMS 2008, les IPSID correspondent aux LMNH de phénotype B de la zone marginale du mucosa-associated lymphoid tissue (MALT) extraganglionnaires. Dans la classification des lymphomes gastro-intestinaux répertoriés par ISAACSON, ils correspondent aux LMNB du MALT de faible degré de malignité, de type méditerranéen (maladie des chaînes ∝essentiellement).
Galian et al. avaient établi des critčres qui classent les IPSID en trois stades selon le grade de malignité [7]. Aux stades A et B, l´intestin gręle est macroscopiquement normal, en dehors d´un épaississement des plis qui sont semés de petits nodules. Au stade C, une ou plusieurs tumeurs ulcérées et/ou un épaississement pariétal étendu sont visibles, des sténoses ou des fistules internes sont possibles.
Sur le plan histologique le stade A se manifeste par une prolifération dense de plasmocytes matures ne franchissant pas la muscularis mucosae. Les villosités sont plus ou moins raccourcies et élargies, le revętement épithélial est peu altéré, les cryptes sont rares, écartées et atrophiques. Le Stade C est caractérisé par la présence d´un lymphome immunoblastique avec différenciation plasmocytaire. Et c'est le cas de notre malade. Au stade B, intermédiaire entre les précédents, l´infiltrat cellulaire, fait de plasmocytes franchement dystrophiques et d´un petit nombre d´immunoblastes (ou centroblastes ) disséminés ou en amas, envahit la sous-muqueuse et parfois la musculeuse.
L´évolution spontanée de l'IPSID peut ętre continue ou, plus fréquemment, interrompue par des périodes d´amélioration clinique parfois induites par un traitement antibiotique aveugle. Le Traitement ŕ un stade précoce(le stade A) fait appel ŕ une bi antibiothérapie ŕ bas de cycline et de métronidazol pour une durée de six mois la durée de rémission peut aller de 36 mois ŕ 5 ans selon une expérience menée ŕ l'hopital de Saint-Lazare, sur la base d'une étude coopérative tuniso-française [11].
Au stade C et au stade B qui ne répondent pas ŕ l'antibiothérapie, une polychimiothérapie incluant une anthracycline (CHOP) est recommandée. En cas de maladie réfractaire ŕ la polychimiothérapie, une chimiothérapie intensive et une auto-greffe sont recommandées. Notre patient avait un bilan d'extension négatif et a été mis sous chimiothérapie ŕ base de CHOP.
Les patients doivent bénéficier d'une évaluation en cours de traitement, ŕ trois et six mois. Elle doit ętre clinique, endoscopique (biopsies jéjunales) et biologique (taux sérique des chaînes lourdes alpha). Il n'existe pas de protocole validé pour la surveillance post-thérapeutique, néanmoins elle doit ętre similaire ŕ celle des autres LMNH (tous les trois mois pendant les deux premičres années, tous les six mois pendant trois ans puis une fois par an).un traitement antibiotique et antiparasitaire doit aussi ętre prescrit, car il peut améliorer le syndrome de malabsorption [11].
Les IPSID sont une maladie orpheline, plusieurs questions demeurent non résolues et méritent des investigations, notamment des études clinicopathologiques étayant le rôle du C. jejuni et la pathogénie de la progression des IPSID.
Les auteurs ne déclarent aucun conflit d'intéręts
Amal Bennani a rédigé le manuscrit et a participé ŕ la prise en charge macroscopique et ŕ l'analyse anatomopathologique de la pičce opératoire. Znati Kaoutar et Affaf Amarti ont également participé au diagnostic anatomopathologique. Salima Rezzouk Hicham Bouhadouti et Khalid Maazaz ont participé ŕ la prise en charge chirurgicale du malade.
Figure 1: Abdomen sans préparation montrant de multiples niveaux hydro-aériques de type gręliques
Figure 2: Scanner abdominal en coupes axiales montrant l’image en cocarde correspondant ŕ l’invagination
Figure 3: Prolifération de cellules de grande taille formant des clusters au sein de cellules de petite taille ŕ différenciation plasmocytaire(HES X 200)
Figure 4: Expression du CD20 par les cellules tumorales
- Vinzio S, Goichot B, Krebs-Wurtz E, Herbrecht R et al. Entéropathie exsudative révélant un lymphome digestif B diffus. La Presse Médicale. Juin 1998; 27(20): 963. PubMed | Google Scholar
- Rambaud JC, Ruskoné-Fourmestraux A. A Small intestina llymphoma. Surv Dig Dis. 1985; 395-113. PubMed | Google Scholar
- Ruskoné-Fourmestraux A, Rambaud JC. Primary gastrointestinal non-Hodgkin's lymphomas.In: P Solal-Celigny, N eds Brousse (Ed) Non-Hodgkin's lymphomasParis : Frison-Roche.1993; 179-191. PubMed | Google Scholar
- Rambaud JC, Halphen M. Immunoproliferative small intestinal disease (IPSID): relationship with alpha chain disease and "Mediterranean " lymphomas.Gastroenterol Int.1989 ; 2 : 33-41. PubMed | Google Scholar
- Lecuit M, Achabin E, Martin A, Royam C, Pochar P, Suarez f, et al. Immunoproliverative small intestinale disease with campylobacter jéjuni. N Engl J Med. 2004 Jan 15;350(3):239-48.. Google Scholar
- M Lopez-Botet, C Chantar, A Plaza, L Abreu, M Kreisler, M O de Landazuri.In vitro immune cell function in six cases of immunoproliferative small intestinal disease after long-term remission.Clin Exp Immunol. 1983 ; 53(3) : 663-671. PubMed | Google Scholar
- Galian A, Lecestre MJ, Scotto J, Bognel C, et al. Pathological study of alpha-chain disease with special emphasis on evolution. Cancer. 1977 May;39(5):2081-101. PubMed | Google Scholar
- Al-Saleem T, Al-Mondhiry H.. Immunoproliferative small intestinal disease (IPSID): a model for mature B-cell neoplasms. Blood. 2005 ; 105(6) : 2274-2280. PubMed | Google Scholar
- Thésaurus National de Cancérologie Digestive.Chapitre : Lymphomes digestifs, version du 03/08/20. Google Scholar
- M L Remedios, M Appleyard. Capsule endoscopy: current indications and future prospects. Int Med J. 2005 ; 35(4) : 234-239. PubMed | Google Scholar
- Ben-Ayed F, Halphen M, Najjar T, Boussene H, Jaadoura H, Bouguerra A , et al.Treatment of alpha chain disease Results of a prospective study in 21 Tunisian patients by the Tunisian-French intestinal lymphoma study group. Cancer. 1989 ; 63(7):1251-6. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
Recently from the PAMJ








Authors´ services
