
Syndrome de Meckel Gruber: ą propos d’un cas rare
Sanaa Itchimouh, Karima Khabtou, Sakher Mahdaoui, Houssine Boufettal, Naima Samouh
Corresponding author: Karima Khabtou, Service de Gynécologie Obstétrique ‘C’, CHU Ibn Rochd, Casablanca, Maroc 
Received: 23 Apr 2016 - Accepted: 08 Aug 2016 - Published: 29 Sep 2016
Domain: Maternal and child health
Keywords: Syndrome de Meckel, dysplasie rénale, encéphalocèle, diagnostic anténatal
©Sanaa Itchimouh et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Sanaa Itchimouh et al. Syndrome de Meckel Gruber: ą propos d’un cas rare. Pan African Medical Journal. 2016;25:43. [doi: 10.11604/pamj.2016.25.43.9696]
Available online at: https://www.panafrican-med-journal.com//content/article/25/43/full
Original article 

Syndrome de Meckel Gruber: ą propos d’un cas rare
Syndrome de Meckel Gruber: ą propos d’un cas rare
Meckel Gruber syndrome: about a rare case
Sanaa Itchimouh1, Karima Khabtou1,&, Sakher Mahdaoui1, Houssine Boufettal1, Naima Samouh1
1Service de Gynécologie Obstétrique ‘C’, CHU Ibn Rochd, Casablanca, Maroc
&Auteur correspondant
Karima Khabtou, Service de Gynécologie Obstétrique ‘C’, CHU Ibn Rochd, Casablanca, Maroc
Le syndrome de Meckel Gruber est un syndrome poly malformatif rare, de transmission autosomique récessive, défini par d'encéphalocčle occipital, polydactylie et dysplasie kystique rénale. L'échographie constitue, ą l'heure actuelle, le meilleur moyen de dépistage anténatal de cette poly malformation létale et sa confirmation se fait par l'étude du caryotype. Nous rapportons un cas de syndrome de Meckel découvert par échographie. La grossesse a été interrompue ą 25 semaines d'aménorrhée.
English abstract
Meckel Gruber syndrome is a rare autosomal recessive polymalformation syndrome characterized by occipital encephalocele, polydactyly and cystic renal dysplasia. Ultrasound is, at present, the best tool for prenatal screening of this lethal polymalformation and diagnosys is confirmed by karyotyping. We here report a case of Meckel Gruber syndrome detected by ultrasound. Abortion was performed at 25 weeks of amenorrhoea.
Key words: Meckel syndrome, renal dysplasia, encephalocele, prenatal diagnosis

Le syndrome de Meckel Gruber décrit par Meckel en 1822, et Gruber en 1934 associant une encéphalocčle, dysplasie kystique des reins, et polydactylie. La variabilité des tableaux cliniques recensés dans la littérature montre que le polymorphisme de ce syndrome est une caractéristique essentielle. L´échographie constitue, ą l´heure actuelle, le meilleur moyen de dépistage anténatal de cette poly malformation létale [1]. Nous rapportons un cas de syndrome de Meckel découvert ą l'échographie sur grossesse de 26 SA+2 jours.

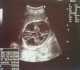
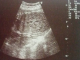
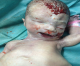
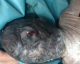
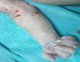
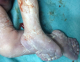
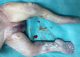
Patiente āgée de 24 ans, primigeste primipare, ayant comme antécédent une consanguinité de premier degré. Elle avait consulté pour la premičre fois pour suivi de grossesse de 26SA + 2 jrs. Une échographie morphologique fœtale a été réalisée objectivant une grossesse monofoetale évolutive avec malformations fœtales comprenant ; hydrocéphalie majeure (Figure 1) + syndrome poly malformatif rénal (Figure 2) et cardiaque associé ą un anamnios sévčre. L'interruption médicale de grossesse a été proposée et acceptée par le couple, elle a abouti ą un accouchement par voie basse d'un fœtus de sexe féminin, poids 1100g. L'examen macroscopique retrouve: au niveau du pole céphalique: rétrognatisme (Figure 3) et encéphalocčle postérieur (Figure 4); au niveau de l'abdomen: une hépato splénomégalie avec ascite; au niveau des membres : une polydactylie (Figure 5) sur les 4 segments distaux, pieds bots (Figure 6) et un aspect incurvé de l'humérus (Figure 7); examen du rachis et les organes génitaux externes est normal. L'examen autopsique a été refusé par la famille. Devant ce syndrome poly malformatif, le diagnostic de syndrome de Meckel a été évoqué.
Définition
Le syndrome de Meckel est un syndrome poly malformatif congénital rare, de transmission autosomique récessive, décrit pour la premičre fois en 1822 dans la littérature allemande. Le syndrome de Meckel est un syndrome héréditaire caractérisé par un ensemble de malformations congénitales touchant notamment le syndrome nerveux central et les reins. Il est généralement fatal peu aprčs la naissance [2].
Epidémiologie
Le syndrome de Meckel touche 1 personne sur 13 250 ą 1 140 000 personnes dans le monde. Plus fréquent en Finlande, oł sa prévalence ą la naissance est de 1 pour 9 000 et oł la fréquence de la mutation est de 1% [3]. Trois gčnes ont été cartographiés: MKS1 sur le chromosome 17, MKS2 sur le 11, et MKS3 sur le 8.
Manifestation clinique
Le syndrome de Meckel est une maladie monogénique caractérisée par la combinaison de kystes rénaux et d'autres manifestations [4]: anomalies de développement du systčme nerveux central (encéphalocčle occipitale); polydactylie; dysplasie des voies biliaires et des kystes hépatiques. Le syndrome de Meckel est défini généralement par la triade: Encéphalocčle occipital, dysplasie kystique des reins et polydactylie. La polydactylie est le plus souvent post-axiale (6čme doigt), mais peut parfois źtre pré-axiale (duplication du pouce) Une incurvation des os longs des membres est présente dans un cas sur 6. D´autres anomalies peuvent źtre présentes: fente labio-palatine , anophtalmie ou une microphtalmie, atrésie de l´určtre, malformations cardiaques et des organes génitaux.
Critčres diagnostiques
critčres majeurs: la dysplasie rénale kystique est un critčre obligatoire pour établir le diagnostic associé ą un anamnios [5-7]; critčres mineurs: la fibrose hépatique; l'encéphalocčle occipitale; polydactylie; autres malformations du systčme nerveux central: malformation de Dandy-Walker et malformation d´Arnold Chiari.
Diagnostic anténatal
Le diagnostic prénatal peut źtre réalisé ą partir d´une image échographique de kyste anéchogčne intracrānien et/ou d´une malformation crānienne ą la fin du premier trimestre ou encore en présence de reins anormalement gros [8]. Les autres caractéristiques du syndrome peuvent źtre décelées plus tardivement ą l´échographie. L´amniocentčse peut révéler un taux élevé d´alpha-foetoprotéine amniotique dū ą l'encéphalocčle [9]. Le caryotype reste le meilleur moyen pour confirmer le diagnostic Si la grossesse est menée ą terme, le nouveau-né décčde en période périnatale.
Evolution
Le syndrome de Meckel est létal avec en moyenne une survie inférieure ą 24 heures. Toutefois Genuardi décrit un cas de syndrome de Meckel comprenant au départ des reins polykystiques, un Dandy-Walker, une polydactylie postaxiale et ayant survécu 43 mois avant de décéder par une insuffisance rénale [10]. Le conseil génétique a pour objectif d´informer les parents d´un individu atteint que le risque de récurrence est de 25% pour les grossesses ultérieures.
Diagnostic différentiel
Les trisomies 13 et 18 sont éliminées devant un caryotype normal [11]. D´autres syndromes poly malformatifs peuvent poser des difficultés diagnostiques plus importantes. Le syndrome de Carpenter-Hunter associe encéphalocčle, dysplasie kystique rénale, polydactylie, mais aussi des lésions osseuses généralisées. On retrouvera aussi la polydactylie dans les syndromes d'Ellis von Creveld, polydactylie cōtes courtes, Moon-Bardet-Biedl, holoprosencéphalie-polydactylie (pseudo-trisomie 13). Une grande aide au diagnostic sera apportée par l´isolement du gčne du syndrome de Meckel.
Conseil génétique
Le syndrome de Meckel est de transmission autosomique récessive. La fréquence du gčne du syndrome de Meckel dans la population générale est de l´ordre de 1/400. A l´heure actuelle, il est encore impossible de dépister précisément le gčne du syndrome de Meckel. Toutefois la localisation récente du locus responsable au niveau du chromosome 17 avec trois gčnes potentiellement impliqués montre que la recherche est proche du but. C´est pourquoi, en l´absence de critčres formellement définis pour le syndrome de Meckel, il est indispensable de conserver des tissus fœtaux pour une analyse génique et moléculaire qui permettra un diagnostic précis. En effet, si dans le cas d´un Dandy Walker isolé le conseil génétique doit źtre rassurant, le risque de récidive locale étant de 1%, en cas de syndrome de Meckel avec Dandy Walker ce risque est de 25% [12].
Les malformations les plus fréquemment retrouvées dans le syndrome de Meckel sont la dysplasie rénale polykystique, l'encéphalocčle, la polydactylie, et la fibrose hépatique, aucune d'entre elles n'est constante. Le polymorphisme de ce syndrome peut źtre considéré comme une caractéristique essentielle, mais qui complique l'accčs ą un diagnostic de certitude. Les progrčs de la génétique avec l'isolement précis du gčne responsable du syndrome de Meckel vont constituer l'étape ultérieure vers un diagnostic de certitude, et son application au diagnostic anténatal.
Les auteurs ne déclarent aucun conflit d’'intérźts.
Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: aspect échographique d’une hydrocéphalie majeure
Figure 2: aspect échographique de kystes rénaux
Figure 3: aspect de rétrognatisme néonatal
Figure 4: encéphalocčle postérieur
Figure 5: polydactylie sur les 4 segments distaux
Figure 6: aspect de pieds bots avec polydactylie
Figure 7: aspect incurvé de l’humérus avec aspect normal des organes génitaux externes
- Tanriverdi HA, Hendrik HJ, Ertan K, Schmidt W. Meckel Gruber syndrome: a first trimester diagnosis of a recurrent case. European Journal of Ultrasound. 2002; 15(1-2):69-72. PubMed | Google Scholar
- Gabrielle Wheway, Zakia Abdelhamed. Aberrant Wnt signalling and cellular over-proliferation in a novel mouse model of Meckel'Gruber syndrome. Developmental Biology. 2013; 377(1): 55-66. PubMed | Google Scholar
- Salonen R. The Meckel syndrome: clinico pathological findings in 67 patients. Am JMed Genet. 1984; 18:671-89. PubMed | Google Scholar
- Nelson J, Nevin NJ, Hanna EJ. Polydactyly in a carrier of the gene for the Meckelsyndrome. Am J Med Genet. 1994; 53(3): 207-9. PubMed | Google Scholar
- Jha T, Bardhan J, Das B, Patra KK, Dhali B, Seth S. Meckel-Gruber syndrome: a rare clinical entity. J Indian Med Assoc. 2010; 108(9): 611-2. PubMed | Google Scholar
- Desai SR, Wader JV. Meckel Gruber Syndrome--a case report. Indian J Pathol Microbiol. 2004; 47(3): 430-2. PubMed | Google Scholar
- Walsh M, Graupman P. Meckle Gruber syndrome in association with occipital meningocele. Pediatr Neurosurg. 2006; 42(5): 333-4. PubMed | Google Scholar
- Van Wymersch D, Favre R. Intérźt de l'échographie tridimensionnelle en obstétrique et gynécologie. Réf Gynécol Obstét. 1995; 31: 82-7. PubMed | Google Scholar
- Ickowicz V, Eurin D, Maugey-Laulom B, Didier F, Garel C, Gubler MC, et al. Meckel-Grüber syndrome: sonography and pathology. Ultrasound Obstet Gynecol. 2006; 27(3): 296-300. PubMed | Google Scholar
- Kheir AEM, Imam A, Omer IM, Hassan IMA, Elamin SA, Awadalla EA et al. Meckel-Gruber Syndrome: a rare and lethal anomaly. Sudan J Paediatr. 2012; 12(1):93-96. PubMed | Google Scholar
- Alexiev BA, Lin X, Sun CC, Brenner DS. Meckel Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med. 2006; 130(8):1236-8. PubMed | Google Scholar
- Philip N, Mattei JF. Malformations congénitales: intérźt génétique et étiologique. Dans Echographie des malformations fœtales. Vigot Paris; 1990 :9-17. Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
PlumX Metrics
Syndrome de Meckel Gruber: ą propos d’un cas rareRecently from the PAMJ








Authors´ services
