
Syndrome de Protée: ŕ propos d’une observation pédiatrique
Kamel Abidi, Manel Jellouli, Abir Boussetta, Yousra Hammi, Tahar Gargah
Corresponding author: Manel Jellouli, Service de Pédiatrie, Hôpital Charles Nicolle de Tunis, Tunisie 
Received: 15 Sep 2015 - Accepted: 12 Oct 2015 - Published: 28 Oct 2015
Domain: Maternal and child health
Keywords: Syndrome de Protée, croissance excessive, complications thromboemboliques
©Kamel Abidi et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Kamel Abidi et al. Syndrome de Protée: ŕ propos d’une observation pédiatrique. Pan African Medical Journal. 2015;22:200. [doi: 10.11604/pamj.2015.22.200.7970]
Available online at: https://www.panafrican-med-journal.com//content/article/22/200/full
Original article 

Syndrome de Protée: ŕ propos d’une observation pédiatrique
Syndrome de Protée: ŕ propos d’une observation pédiatrique
Kamel Abidi1, Manel Jellouli1,&, Abir Boussetta1, Yousra Hammi1, Tahar Gargah1
1Service de Pédiatrie, Hôpital Charles Nicolle de Tunis, Tunisie
&Auteur correspondant
Manel Jellouli, Service de Pédiatrie, Hôpital Charles Nicolle de Tunis, Tunisie
Le syndrome de Protéeest une maladie trčs rare, se traduisant par une croissance excessive pouvant toucher différents organes et tissus de l'organisme. Ses complications sont dominées par les complications thromboemboliques et tumorales. Nous rapportons le cas d'un malade suivi pour un syndrome de Protée qui présentait un gigantisme des membres supérieurs avec une macrodactylie et une hypertrophie du thorax ainsi qu'une scoliose.
Le syndrome de Protée est une maladie génétique complexe comprenant des hamartomes de taille importante impliquant plusieurs tissus: tissu conjonctif, tissu épidermique et tissu osseux. Il se manifeste dčs la naissance et les hamartomes grandissent au cours de la vie. C'est un syndrome rare associant un surdéveloppement local (allant de la macrodactylie ŕ l'hémihypertrophie), des tumeurs de sičge variable et des anomalies vasculaires. Il s'agit essentiellement de malformations capillaires et veineuses mais des malformations lymphatiques sont associées [1]. Nous rapportons ici le cas d'un enfant âgé de dix ans, suivi dans notre service pour un syndrome de Protée.
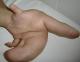
Il s'agit d'un patient de dix ans, suivi depuis l'âge de deux ans pour un syndrome polymalformatif. Cet enfant, sans antécédents familiaux notables, est né de parents consanguins de deuxičme degré. L'examen clinique a retrouvé un gigantisme des membres supérieurs avec une macrodactylie et une hypertrophie du thorax ainsi qu'une scoliose.Il n'existait pas d'hypertrophie des organes génitaux externes.Par ailleurs, il avait un bon développement psychomoteur, L'examen cardiovasculaire ainsi que l'examen pleuro-pulmonaire étaient sans anomalies.L'examen ophtalmologique n'a pas montré d'anomalie, ainsi que l'échographie doppler abdominal.Les radiographies du squelette ont révélé une hypertrophie osseuse.Le syndrome de Protée a été porté devant l'association d'une hypertrophie des membres supérieurs et du thorax ainsi qu'une scoliose (Figure 1, Figure 2). Ce patient a été hospitalisé ŕ plusieurs reprises pour érysipčle des membres supérieurs et pour thrombose des veines superficielles des membres supérieurs, avec une évolution clinique favorable.
Le syndrome de Protée est une maladie congénitale trčs rare qui induit une croissance excessive et déformante de multiples tissus du corps notamment conjonctif, osseux et adipeux avec une distribution en mosaďque des lésions [2]. Le plus célčbre patient atteint de syndrome de Protée est John Merrick alias « Elephant Man » longtemps considéré ŕ tort comme porteur d'une neurofibromatose. Temtamy et Rogers sont les premiers ŕ avoir décrit le syndrome de Protée en 1976 [3]. Ce sont Wiedman et al. [4] qui étaient les premiers ŕ lui donner le nom de syndrome de Protée d'aprčs le dieu grec Protée qui avait la faculté de changer de forme pour échapper ŕ ses ennemis. Il s'agit d'une pathologie exceptionnelle. Turner et al. [2] dans leur revue systématique avaient recensé en 2004, 205 cas publiés dans la littérature internationale. Parmi ces cas, seuls 97 étaient réellement des SP si les critčres diagnostiques de Biesecker et al étaient respectés. Ceci souligne la rareté et la difficulté diagnostique de la pathologie. Le diagnostic de SP repose sur l'association de critčres généraux et spécifiques [5] qui sont résumés dans le Tableau 1. De nombreux diagnostics différentiels peuvent se poser avec d'autres pathologies entraînant une hypertrophie corporelle telle que le syndrome de la lipomatose multiple/hémihyperplasie, la lipomatose encéphalo-cranio-cutanée ou encore la maladie de Klippel-Trenaunay [6]. Sur le plan clinique, la maladie se manifeste par une mégalodactylie, des anomalies vertébrales, une croissance asymétrique des membres avec une inégalité de longueur, des hyperostoses, un développement excessif des viscčres ; rate et thymus en particulier. Une répartition anormale et disproportionnée des graisses ainsi qu'un développement asymétrique des muscles, des nćvi du tissu conjonctif et des nćvi épidermiques font partie du tableau clinique de la maladie. Certains malades peuvent présenter des malformations vasculaires. L'incidence des manifestations thromboemboliques ainsi que des malformations pulmonaires kystiques est élevée.
Dans notre cas, le patient présente un gigantisme des membres supérieurs avec hypertrophie des doigts et du thorax, une scoliose et une hyperostose.Il a présenté plusieurs épisodes de thromboses sur des malformations artério veineuses.Les manifestations cutanées étaient ŕ type d'érysipčle récidivant et d'abcčs secondaires aux épisodes itératifs de thromboses des malformations vasculaires. Les manifestations extra cutanées se traduisent par une croissance squelettique et viscérale importante, ainsi que par des tumeurs, des kystes, des malformations vasculaires. Tous les malades développent habituellement une croissance exagérée et disproportionnée des mains et des pieds ainsi que des doigts aboutissant ŕ une macrodactylie. Les lipomes sont les tumeurs les plus fréquemment rencontrées au cours du syndrome de Protée. D'autres tumeurs sont néanmoins plus spécifiques de la maladie ŕ savoir les tumeurs ovariennes bilatérales chez les filles, les adénomes parotidiens monomorphes qui se voient surtout aprčs l'âge de vingt ans.Ces malades peuvent développer aussi des méningiomes, des tumeurs testiculaires, du nerf optique, et des seins avec la possibilité de survenue concomitante de plusieurs types de ces tumeurs.Dans notre cas, aucune manifestation tumorale n'a été observée.
Sur le plan génétique, le SP est dű ŕ une mutation somatique en mosaďque activatrice de l'oncogčne AKT1 porté par des cellules somatiques expliquant la survenue souvent sporadique et la répartition des lésions de maničre disséminée et asymétrique [7]. Des mutations du gčne PTEN (retrouvés dans le syndrome de Bannayan-Zonana), ont été retrouvées sur la lignée germinale de certains patients suivis pour un syndrome de Protée mais n'ont pas été confirmées dans la majorité des cas.Une mutation génétique, n'a pu ętre retrouvée chez notre malade [8].
Le syndrome de Protée est une cause fréquente de malformations artério-veineuse pouvant se surinfecter fréquemment. Les autres complications sont dominées par les tumeurs et les complications orthopédiques qui peuvent mettre en jeu le pronostic fonctionnel et męme vital des malades.
Les auteurs ne déclarent aucun conflit d'intéręt.
Tous les auteurs ont contribué ŕ l'élaboration de ce travail. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Tableau 1: critčres diagnostiques de Biesecker
Figure 1: macrodactylie
Figure 2: gigantisme des
membres supérieurs avec et hypertrophie du thorax
- Li CY, Chang YL, Chen WC et al. Pulmonary manifestations and management of Proteus syndrome. J Formos Med Assoc. 2010; 109(5):397-400. PubMed | Google Scholar
- Turner JT, Cohen Jr MM, Biesecker LG. Reassessment of the Proteus syndromeliterature: application of diagnostic criteria to published cases. Am J Med Genet. 2004;130 (2):111-22. PubMed | Google Scholar
- Cohen Jr MM. Understanding Proteus syndrome, unmasking the elephant man, and stemming elephant fever. Neurofibromatosis. 1988; 1 (5-6):260-80. PubMed | Google Scholar
- Temtamy SA, Rogers JG. Macrodactyly, hemihypertrophy, and connective tis-sue nevi: report of a new syndrome and review of the literature. J Pediatr.1976; 89 (6):924-7. PubMed | Google Scholar
- Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham Jr JM, ViljoenDL et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patientevaluation. Am J Med Genet. 1999;84 (5):389-95. PubMed | Google Scholar
- Cohen Jr MM. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137 (1):38-52. PubMed | Google Scholar
- Lindhurst MJ, Sapp JC, Teer JK et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011; 365 (7):611-9. PubMed | Google Scholar
- Boccone L, Dessi V, Serra G et al. Bannayan-Riley-Ruvalcaba syndrome with posterior subcapsular congenital cataract and a consensus sequence splicing PTEN mutation. Am J Med Genet. 2008; 146 (2):257-60. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
Recently from the PAMJ








Authors´ services
