
L’ostéogenčse imparfaite: ŕ propos d’un cas
Najeh Hsayaoui, Chaouki Mbarki, Saoussen Melliti, Youssef Elkadhi, Fatma Douik, Sana Mezghanni, Hedhili Oueslati
Corresponding author: Mbarki Chaouki, Service de Gynécologie Obstétrique Hôpital Ben Arous, Tunis, Tunusie 
Received: 23 Feb 2015 - Accepted: 11 May 2015 - Published: 03 Jun 2015
Domain: Maternal and child health
Keywords: Ostéogenèse imparfaite, échographie, diagnostic
©Najeh Hsayaoui et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Najeh Hsayaoui et al. L’ostéogenčse imparfaite: ŕ propos d’un cas. Pan African Medical Journal. 2015;21:83. [doi: 10.11604/pamj.2015.21.83.6416]
Available online at: https://www.panafrican-med-journal.com//content/article/21/83/full
Original article 

L’ostéogenčse imparfaite: ŕ propos d’un cas
L’ostéogenčse imparfaite: ŕ propos d’un cas
Najeh Hsayaoui1, Chaouki Mbarki1,&, Saoussen Melliti2, Youssef El Cadhi1, Fatma Douik1, Sana Mezghanni2, Hedhili Oueslati1
1Service de Gynécologie Obstétrique Hôpital Ben Arous, Tunis, Tunusie, 2Service de Radiologie Hôpital Ben Arous, Tunis, Tunusie
&Auteur correspondant
Mbarki Chaouki, Service de Gynécologie Obstétrique Hôpital Ben Arous, Tunis, Tunusie
L'ostéogenčse imparfaite est une maladie héréditaire caractérisée par une fragilité osseuse secondaire ŕ un défaut de synthčse du collagčne de type I. Le diagnostic est suspecté devant des signes échographiques évocateurs et confirmé par une étude génétique. Nous rapportons un cas d'ostéogenčse imparfaite de découverte tardive au troisičme trimestre chez une patiente qui n'a pas suivi sa grossesse.
L'ostéogenčse imparfaite (OI) est une maladie héréditaire caractérisée par une fragilité osseuse secondaire ŕ un défaut de synthčse du collagčne de type I [1]. Le tableau clinique est variable pouvant inclure des fractures prénatales et décčs périnatal, ou des formes trčs frustes. Des troubles extrasquelettiques peuvent ętre associés ŕ des degrés variables. C'est une maladie rare qui touche hommes et femmes, sans prédominance ethnique ou raciale [2].
Mme H.Z est âgée de 26 ans. C'est une deuxičme geste. Elle a un enfant en bon état de santé apparente. Il n'y a pas de notion de mariage consanguin. Mme H.Z. n'a pas suivi sa grossesse en cours. Elle a consulté pour la premičre fois ŕ 32 SA pour échographie obstétricale. Cet examen a révélé un foetus malformé présentant un nanisme micromélique associant des membres courts et incurvés avec fracture déplacée du fémur droit, le crâne est allongé (Figure 1). Le diagnostic d'OI a été fortement évoqué. La patiente a eu une corticothérapie anténatale avec accouchement par césarienne ŕ 38 SA. L'examen néonatal a objectivé une déformation de la cuisse droite en rapport avec la fracture pathologique survenue « in utéro ». La radiographie du squelette a montré un défaut majeur de l'ossification touchant tous les os (Figure 2) associé ŕ des déformations des os des membres (avant-bras, cuisses et jambes). L'enfant a survécu pendant une année. Au cours de cette période, il a eu des multiples fractures des membres supérieures et inférieures et il est décédé ŕ l'âge de un an dans un tableau de détresse respiratoire.
L'ostéogenčse imparfaite fait partie d'un groupe d'affections héréditaires ŕ transmission dominante, caractérisée par une anomalie de la synthčse du collagčne de type 1 (production réduite et collagčne anormal). C'est une maladie rare. Elle atteint un nouveau-né sur 25 ŕ 50.000, sans prédominance de sexe ou de race et sans distribution géographique préférentielle [1]. Les fractures multiples par fragilité osseuse représentent la principale manifestation de la maladie, et sont responsables de la morbidité et la mortalité de l'ostéogenčse imparfaite par insuffisance respiratoire secondaire aux fractures costales [3]. Des troubles extra squelettiques sont associés ŕ des degrés variables avec retentissement plus ou moins important. Une laxité ligamentaire qui peut compromettre la stabilité articulaire, des troubles oculaires et auditives, des anomalies cardio-vasculaires, des troubles de l'hémostase, des affections rénales et une dentinogenčse imparfaite sont les manifestations les plus fréquentes [1,2].
La classification de Sillence et al [4], complétée en 2004 par Rauch et Glorieux [5], classiquement la plus employée, distingue actuellement sept formes. En dehors des formes de sévérité extręme, il peut ętre difficile de classer un patient lors de la découverte de la pathologie, surtout dans les premičres années de vie. Il peut donc ętre utile d'utiliser une classification selon l'âge d'apparition [6].
Le diagnostic anténatal est évoqué généralement devant des anomalies osseuses ŕ type de fracture, incurvation, déformation ou raccourcissement. Rarement une déformabilité du crâne sous le passage de la sonde d'échographie peut attirer l'attention. Le pronostic est difficile ŕ établir précocement. L'évolution est imprévisible. Des fractures multiples et des déformations osseuses peuvent se voir avec retentissement statural et respiratoire variable [1]. Le diagnostic de l'OI dans ses formes ŕ révélation postnatale est clinique, reposant sur les manifestations squelettiques décrites ci-dessus ainsi que les manifestations extra squelettiques. Les différentes manifestations extra squelettiques sont variables. On peut noter une laxité ligamentaire avec retentissement variable sur la statique, une coloration bleutée des sclérotiques due ŕ la transparence excessive de la sclérotique, une perte de l'audition, une atteinte du systčme cardiovasculaire. Les ecchymoses et les épistaxis sont fréquentes chez l'enfant atteint d'ostéogenčse imparfaite ainsi que les atteintes rénales neurologiques et hépatiques [2].
Bien que le diagnostic de l'OI est basé sur la clinique et l'échographie, les progrčs de la génétique moléculaire ont permis de localiser les gčnes responsables de la maladie. Il s'agit de deux gčnes, Col 1 A1 situé sur le bras long q du chromosome 17 entre les positions 21,31 et 22,15, et Col 1 A2 situé sur le bras long q du chromosome 7 entre les positions 21,3 et 22, 1. Ces deux gčnes Col 1 A1 et Col1 A2 codent respectivement pour la synthčse des chaînes a1 et a2 du collagčne de type I. Ces mutations sont pour la plupart dominantes, elles sont soit de novo, ce qui est le plus fréquent, soit résultent d'une mosaďque d'un des parents [7,8].
Le traitement de cette affection est symptomatique. Le pronostic fonctionnel dépend de la sévérité de l'atteinte et de sa prise en charge. L'utilisation récente des bisphosphonates, associée ŕ la stimulation motrice et ŕ la chirurgie, a beaucoup amélioré l'autonomie des sujets ayant une forme grave. Le pronostic vital est lié ŕ l'atteinte respiratoire corrélée ŕ la sévérité des déformations rachidiennes [2].
Le diagnostic d'une ostéogenčse imparfaite doit etre évoqué échographiquement devant des déformations des membres. Un conseil génétique doit ętre proposé en cas d'antécédent d'une ostéogenčse imparfaite dans la famille.
Les auteurs ne déclarent aucun conflit d'intéręt.
Tous les auteurs ont contribué ŕ la rédaction de ce manuscrit lu et approuvé la version finale.
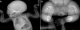
Figure 1: image échographique
de l’ostéogenčse imparfaite; (A) fracture déplacée du fémur, (B) aspect
allongé du crane
Figure 2: radiographie
du squelette aprčs la naissance; (A) défaut d’ossification du crâne, membres
supérieures trčs courts, côtes déformées porteuses de cals, (B) membres inférieures
courts et incurvés
- Baujat G, Lebre A-S, Cormier-Daire V et al. Ostéogenčse imparfaite, annonce du diagnostic (classification clinique et génétique). Archives de Pédiatrie. 2008;15(5):789-791. PubMed | Google Scholar
- Forin V. Ostéogenčse imparfaite. Press Med. 2007 Dec;36(12 Pt 2):1787-93. PubMed | Google Scholar
- Coutouly X, Bibi R, Magni C. Dissection isolée de l'artčre basilaire dans un cas d'ostéogénčse imparfaite. J Radiol. 2005;86(1):86-8. PubMed | Google Scholar
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101-16. PubMed | Google Scholar
- Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377-85. PubMed | Google Scholar
- Maroteaux P, le Merrer M. Osteogenesis Imperfecta. In: Flammation des maladies osseuses de l'enfant. Paris: Médecine-Sciences. 2002; 4e édition:184-93. PubMed | Google Scholar
- Redon JY, Glaguen D, Collet M. L'ostéogenčse imparfaite: réflexion sur le diagnostic prénatal. J Gynecol Obstet Biol Report. 1993;22(2):173-8. PubMed | Google Scholar
- Raghunath M, Mackay K, Dalgleish et al. Genetic counselling on brittle grounds: recurring osteogenesis imparfecta due to parental mosaicism for a dominant mutation. Eur J Pediatr. 1995;154(2):123-9. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
PlumX Metrics
L’ostéogenčse imparfaite: ŕ propos d’un casRecently from the PAMJ








Authors´ services
