
Grande tache pigmentťe pileuse rťvťlant une forme familiale de la maladie de Von Recklinghausen
Anass Es seddiki, Sahar Messaoudi, Dounia Jaafar, Anass Ayyad, Rim Amrani
Corresponding author: Anass Es seddiki, Service de Pťdiatrie, CHU Mohamed VI, Oujda, Maroc 
Received: 22 Feb 2015 - Accepted: 15 May 2015 - Published: 08 Jul 2015
Domain: Clinical medicine
Keywords: Neurofibromatose 1, grande tache cutanée pigmentée pilleuse, nodules de Lisch
©Anass Es seddiki et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Anass Es seddiki et al. Grande tache pigmentťe pileuse rťvťlant une forme familiale de la maladie de Von Recklinghausen. Pan African Medical Journal. 2015;21:188. [doi: 10.11604/pamj.2015.21.188.6406]
Available online at: https://www.panafrican-med-journal.com//content/article/21/188/full
Original article 

Grande tache pigmentťe pileuse rťvťlant une forme familiale de la maladie de Von Recklinghausen
Grande tache pigmentťe pileuse rťvťlant une forme familiale de la maladie de Von Recklinghausen
Anass Es seddiki1,&, Sahar Messaoudi1, Dounia Jaafar2, Anass Ayyad1, Rim Amrani1
1Service de Pťdiatrie, CHU Mohamed VI, Faculté de Médecine et de Pharmacie d’Oujda, Université Mohammed 1er, Oujda, Maroc, 2Service díOphtalmologie, CHU Mohamed VI, Faculté de Médecine et de Pharmacie d’Oujda, Université Mohammed 1er, Oujda, Maroc
&Auteur correspondant
Anass Es seddiki, Service de Pťdiatrie, CHU Mohamed VI, Faculté de Médecine
et de Pharmacie d’Oujda, Université Mohammed 1er, Oujda, Maroc
La neurofibromatose de type 1 ou maladie de Von Recklinghausen appartient au groupe de maladies appelťes phacomatoses. C'est une affection autosomique dominante relativement rare. La NF1 est caractťrisťe par une extrÍme variabilitť clinique que l'on retrouve ťgalement au sein d'une mÍme famille. Le tableau clinique de la NF1 associe, le plus souvent, de multiples taches cafť au lait, des lentigines axillaires ou inguinales, des neurofibromes cutanťs et des nodules de Lisch. Les difficultťs d'apprentissage sont frťquentes et peuvent Ítre graves dans certaines formes cliniques. Il est important de dťtecter prťcocement les neurofibromes plexiformes, les gliomes intracťrťbraux, les tumeurs des gaines nerveuses, les anomalies vasculaires et les dysplasies osseuses. L'ťvolution est imprťvisible ce qui rend le pronostic incertain par une ťventuelle survenue de dťgťnťrescence malignes. Nous rapportons ici l'observation d'une grande tache cutanťe pigmentťe pilleuse de dťcouverte fortuite qui nous a rťvťlť deux cas familiaux de NF1 d'expression diffťrente.
La maladie de Von Recklinghausen ou la neurofibromatose de type 1 (NF1) est une affection autosomique dominante, elle est caractťrisťe par une extrÍme variabilitť clinique au sein d'une mÍme famille [1,2]. Le tableau clinique de la NF1 associe, le plus souvent, de multiples taches cafť au lait, des lentigines axillaires et inguinales, des neurofibromes cutanťs et des nodules de Lisch. Les difficultťs d'apprentissage sont frťquentes, il est important de dťtecter prťcocement les neurofibromes plexiformes, les gliomes intracťrťbraux, les tumeurs des gaines nerveuses, les anomalies vasculaires et les dysplasies osseuses [1-3]. Nous rapportons ici l'observation de deux cas familiaux de neurofibromatose 1 (NF1) ou maladie de Von Recklinghausen dťcouverts exceptionnellement et fortuitement par une grande tache pigmentťe pileuse.
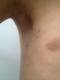
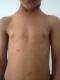
Il s'agit d'une fille ‚gťe de cinq ans, seconde d'une fratrie de 2, de parents consanguins de premier degrť, qui consultait pour un simple ťpisode d'angine, au cours de l'examen clinique on note la prťsence d'une grande tache cutanťe pigmentťe pilleuse lťgŤrement infiltrťe du bras gauche d'allure congťnitale (Figure 1) et quelques taches lentigineuses (lentigines) axillaires (Figure 2). L'examen ophtalmologique sous lampe ainsi qu'un fond d'oeil ťtaient normaux. Une TDM cťrťbrale ťtait normale. La biopsie de la tache pileuse ťtait en faveur d'un neurofibrome plexiforme. Un bilan standard comportant un hťmogramme et ionogramme (fonction rťnale, hťpatique et thyroÔdienne) ťtait normal.
†
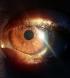
L'enquÍte auprŤs de la famille retrouve des difficultťs d'apprentissage et des ťchecs scolaires chez le frŤre ‚gť de 10 ans, dont l'examen cutanťo-muqueux retrouve des taches cutanťes cafť au lait (plus de 10) au niveau de l'abdomen, du dos, du cou et des deux bras (Figure 3) et quelques lentigines axillaires. A l'examen ophtalmologique sous lampe ŗ fente, on retrouve plusieurs nodules de Lisch (ou hamartomes de l'iris) au niveau des deux yeux (Figure 4) avec un fond d'oeil normal sans trouble visuel. Une TDM cťrťbrale ťtait normale. Un bilan standard comportant un hťmogramme et un ionogramme ťtait normal. Les parents ne prťsentaient aucune atteinte cutanťe ou ophtalmique. Une surveillance hťmatologique, dermatologique et ophtalmologique est programmťe tous les six mois pour nos deux patients.
La neurofibromatose de type 1 (NF1) ou maladie de Von Recklinghausen appartient au groupe de maladies appelťes phacomatoses ou dysplasie ectodermique congťnitale ou encore syndrome neurocutanť congťnital et elle reprťsente 95 % des neurofibromatoses. Elle est relativement rare, sa prťvalence est d'environ 1/4000 ŗ 1/3000 avec une rťpartition mondiale homogŤne et une incidence estimťe ŗ 1/2500 naissances [1,3,4].
†
C'est une affection autosomique dominante et sa pťnťtrance est quasi complŤte ŗ l'‚ge de huit ans. Le gŤne NF1, responsable de la maladie, est localisť sur le bras long du chromosome 17 en 17q11.2 (isolť en 1990) ; il code pour une protťine, la neurofibrine, qui a un rŰle oncosuppresseur [1,2]. L'inactivation de ce gŤne expliquerait l'apparition de nombreux types de tumeurs au cours de la maladie. Selon la confťrence de consensus de 1988 du National Institute of Health Development., le diagnostic de la maladie repose sur la prťsence d'au moins deux des critŤres suivants [1,2,4]: au moins six taches cafť au lait de plus de 5 mm avant la pubertť et plus de 15 mm aprŤs la pubertť ; deux neurofibromes ou plus, un neurofibrome plexiforme ou plus ; des taches lentigineuses de la rťgion axillaire ou inguinale ; deux nodules de Lisch (hamartomes de l'iris) ou plus, un gliome des voies optiques ; une lťsion osseuse caractťristique (pseudarthrose d'un os long, dysplasie sphťno-orbitaire ou cyphose cervicale) ; un parent du premier degrť atteint de NF1 suivant les critŤres prťcťdents.†
Pour notre patiente le diagnostic de la NF1 a ťtť retenu devant la prťsence d'un neurofibrome plexiforme, des lentigines axillaires et les critŤres retrouvťs chez le frŤre : plus de 10 taches cafť au lait, lentigines axillaires et plusieurs nodules de Lisch ŗ l'examen ophtalmologique confirmant ainsi la variabilitť de cette pathologie au sein d'une mÍme famille.†
Les neurofibromes plexiformes se dťveloppent sur le trajet des troncs nerveux des diffťrents plexus et leurs branches de division ŗ l'ťtage cr‚nio-facial, thoracique et abdomino-pelvien, [1,5]. Ce sont des tumeurs bťnignes constituťes de fibroblastes, de cellules axonales, de cellules de Schwann, et de mastocytes, au sein d'une matrice myxoÔde et de fibres de collagŤnes [5]. Le plus souvent, ces neurofibromes simulent des adťnopathies et orientent vers divers diagnostics diffťrentiels (tuberculose, lymphome, sarcoÔdose ou abcŤs) [5]. Le neurofibrome plexiforme se manifeste rarement par une grande tache cutanťe pigmentťe pilleuse au cours de la NF1 (diagnostic ŗ tort d'un naevus de Becker ou du naevus congťnital pileux), c'est une forme exceptionnelle est peu dťcrite dans la littťrature chez l'enfant; un cas rapportť par Schaffer J-V et al (USA) en 2007 et 4 cas pťdiatriques rapportťs par Poiraud C et al (France) en 2011 [6,7].†
Pendant l'enfance, les complications de la NF1 sont dominťes par le gliome des voies optiques ; 15 % (indication de l'IRM cťrťbrale), des troubles neuropsychologiques (les difficultés cognitives retrouvées dans 30 à 65 % des cas) et les complications orthopťdiques (30 %), alors que chez l'adulte les complications esthťtiques et le risque de dťgťnťrescence tumorale sont au premier plan [2,4,8]. Chaix Y explique en 2004 que les troubles cognitifs au cours de la NF1 (comme le cas du frŤre) surviennent indťpendamment de l'existence ou non de lťsions cťrťbrales. Les souris porteuses d'une mutation ŗ l'ťtat homozygote d'un exon situť dans le GTP-Activating Protein Related Domain de la neurofibromine ne prťsentent pas de prťdisposition particuliŤre pour les tumeurs mais dťveloppent des troubles des apprentissages [9].†
Dans un projet de corrťlation gťnotype-phťnotype rťalisť en France entre 2003 et 2005 chez 1099 patients suivis pour NF1, les chercheurs ont confirmť l'hypothŤse du rŰle hormonal dans l'apparition et le dťveloppement des neurofibromes aprŤs la pubertť et en post-partum (dťmontrť in vitro par prolifťration des cellules de Schwann sous l'influence de diffťrentes hormones stťroÔdiennes) et ils ont suggťrť ťgalement l'existence d'une pťriode ŗ risque de dťvelopper des neurofibrome (entre 15 et 30 ans) liťe ŗ la stimulation hormonale [10].†
Les neurofibromes peuvent dťgťnťrer dans 3 ŗ 15 % des cas en neurofibrosarcomes. Le risque de dťgťnťrescence justifie une exťrŤse systťmatique surtout devant une tumeur douloureuse [4,8,11]. Des essais de prise en charge thťrapeutique par biologie molťculaire de certaines voies de signalisation (Ras, mTor) ont ťtaient rťalisť par Valeyrie-Allanore L et al en 2012 pour des patients NF1 porteurs d'une tumeur maligne des gaines nerveuses ou d'un neurofibrome plexiforme, ils ont conclus ŗ des effets prťliminaires encourageants par le Sirolimus (inhibiteurs de mTor) versus Sorafťnib [12].
Cette observation s'ajoute au cas pťdiatriques de NF1, dťcouverte fortuitement par une grande tache cutanťe pigmentťe pilleuse. Une TDM ou IRM cťrťbrale, un examen dermatologique et ophtalmologique et un bilan hťmatologique doivent Ítre rťalisťs devant tout neurofibrome plexiforme surtout chez l'enfant. Le risque de dťgťnťrescence sarcomateuse justifie l'exťrŤse de la lťsion aussi complŤte que possible. Le suivi de ce type de patients doit Ítre assurť par une ťquipe pluridisciplinaire avec surveillance annuelle clinique et radiologique jusqu'ŗ l'‚ge de dix ans, puis rťguliŤre, pour ťvaluer une ťventuelle rťcidive ou une transformation maligne.
Les auteurs ne dťclarent aucun conflit d'intťrÍt.
Tous les auteurs ont lu et approuvť la version finale de ce manuscrit.
Figure 1: grande tache cutanťe pigmentťe pilleuse lťgŤrement infiltrťe du bras gauche chez notre patiente
Figure 2: plusieurs taches lentigineuses chez notre patiente
Figure 3: plusieurs taches cutanťes cafť au lait chez le frŤre
Figure 4: nodules de Lisch (hamartomes de líiris) retrouvťs chez le frŤre
- Pinson S, Wolkenstein P. La neurofibromatose 1 (NF1) ou maladie de Von Recklinghausen. La revue de mťdecine interne. 2005;26(3):196-215. PubMed | Google Scholar
- Rodriguez D. Diagnostic et prise en charge globale des enfants atteints de neurofibromatose de type 1. Arch pťdiatr. 2004;11(6):545-547. PubMed | Google Scholar
- Jacques C, Dietemann J-L. Imagerie de la neurofibromatose de type 1. J Neuroradiol. 2005;32(3):180-197. PubMed | Google Scholar
- Pinson S, Crťange A, Barbarot S, Stalder J-F, Chaix Y, Rodriguez D et al (rťseau NF-France). Neurofibromatose 1: recommandations de prise en charge. Arch Pťdiatr. 2002;9(1):49-60. PubMed | Google Scholar
- Benaissa L, El Kharras A, Bassou D et al. Masse cervicale gauche. Feuillets de Radiologie. 2009;49(3):207-210. PubMed | Google Scholar
- Poiraud C, Barbarot S, Frot A-S, Stalder J-F. Grandes taches pigmentťes pileuses au cours de la neurofibromatose de type 1 de l'enfant: une forme clinique de neurofibrome plexiforme. Annales de Dermatologie et de Vťnťrťologie. 2011;138(12):A149. PubMed | Google Scholar
- Schaffer J-V, Chang M-W, Kovich O-I, Kamino H, Orlow S-J. Pigmented plexiform neurofibroma: distinction from a large congenital melanocytic nevus. Journal of the American Academy of Dermatology. 2007;56(5):862-868. PubMed | Google Scholar
- Landrieu P. Comment surveiller un enfant atteint de neurofibromatose de Recklinghausen? Arch Pťdiatr. 2002;9(8):771-3. PubMed | Google Scholar
- Chaix Y. Apports de la recherche dans la comprťhension des troubles des apprentissages dans la neurofibromatose de type 1. Arch Pťdiatr. 2004;11(6):548-549. PubMed | Google Scholar
- Sbidian E , Duong T-A, Valeyrie-Allanore L , Sabbagh A. Neurofibromatose 1: influence des phťnotypes fťminin et masculin. Annales de Dermatologie et de Vťnťrťologie. 2011;138(12 suppl):A77-A78. PubMed | Google Scholar
- Halb C, LefŤbvre F, Chaouadi D, Munzer M, Behar C. Neurofibromatose de type 1: 3 nouveaux cas de dťgťnťrescence sarcomateuse. Arch Pťdiatr. 2008;15(5):887-922. PubMed | Google Scholar
- Valeyrie-Allanore L, Combemale P, Pouessel D, Culine S, Hamel-Teillac D, Perel Y et al. Thťrapeutiques ciblťes dans la neurofibromatose 1: les premiŤres avancťes. Annales de Dermatologie et de Vťnťrťologie. 2012;139(12 suppl):B104. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ





Authors´ services
