
A propos d’une forme rare de la myosite ossifiante progressive de Munchmeyer
Hicham Sator, Rachida Dafiri, Latifa Chat
Corresponding author: Hicham Sator, Hôpital des Enfants, Service de Radiologie, CHU Ibn Sina, Rabat, Maroc 
Received: 29 May 2014 - Accepted: 10 Mar 2015 - Published: 28 Apr 2015
Domain: Clinical medicine
Keywords: Congénital, enfant, myosite, ossification hétérotopique, radiologie
©Hicham Sator et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hicham Sator et al. A propos d’une forme rare de la myosite ossifiante progressive de Munchmeyer. Pan African Medical Journal. 2015;20:416. [doi: 10.11604/pamj.2015.20.416.4716]
Available online at: https://www.panafrican-med-journal.com//content/article/20/416/full
Original article 
A propos d’une forme rare de la myosite ossifiante progressive de Munchmeyer
A propos d’une forme rare de la myosite ossifiante progressive de Munchmeyer
Hicham Sator1,&, Rachida Dafiri1, Latifa Chat1
1Hôpital des Enfants, Service de Radiologie, CHU Ibn Sina, Rabat, Maroc
&Auteur correspondant
Hicham Sator, Hôpital des Enfants, Service de Radiologie, CHU Ibn Sina, Rabat, Maroc
La myosite ossifiante progressive est une maladie génétique rare. On rapporte le cas d'un jeune garçon de 10 ans qui présentait de multiples tuméfactions douloureuses d'apparition spontanée et progressive au niveau du dos et des membres supérieurs. Ces tuméfactions étaient associées ŕ une fébricule et un hallux valgus bilatéral. Les aspects radiologiques et tomodensitométriques étaient largement suffisants pour confirmer le diagnostic. Le traitement était purement médical. L'évolution était marquée par l'apparition d'autres ossifications des fascias et des muscles aboutissant ŕ des raideurs articulaires trčs invalidantes. Les circonstances de découverte, les aspects épidémiologiques, étiopathogénique, diagnostique, évolutive et thérapeutique sont discutées ŕ travers une revue de la littérature.
La myosite (ou fibrodysplasie) ossifiante progressive (MOP ou FOP), encore appelée maladie de MUNCHMEYER est une maladie génétique extręmement rare ŕ transmission autosomique dominante et ŕ expression variable, caractérisée par des malformations congénitales des gros orteils et une ossification hétérotopique progressive des tissus musculaires et conjonctifs siégeant surtout au niveau de la région cervicale et dorsale, qui évolue par poussées et qui mčne ŕ des raideurs articulaires invalidantes. Les patients se voient comme enfermés dans un «deuxičme squelette». Nous illustrons cette maladie par l'observation clinique d'un petit garçon de 10 ans afin d'en montrer les circonstances de découverte, les aspects épidémiologique, étiopathogénique, diagnostique, évolutive et thérapeutique.
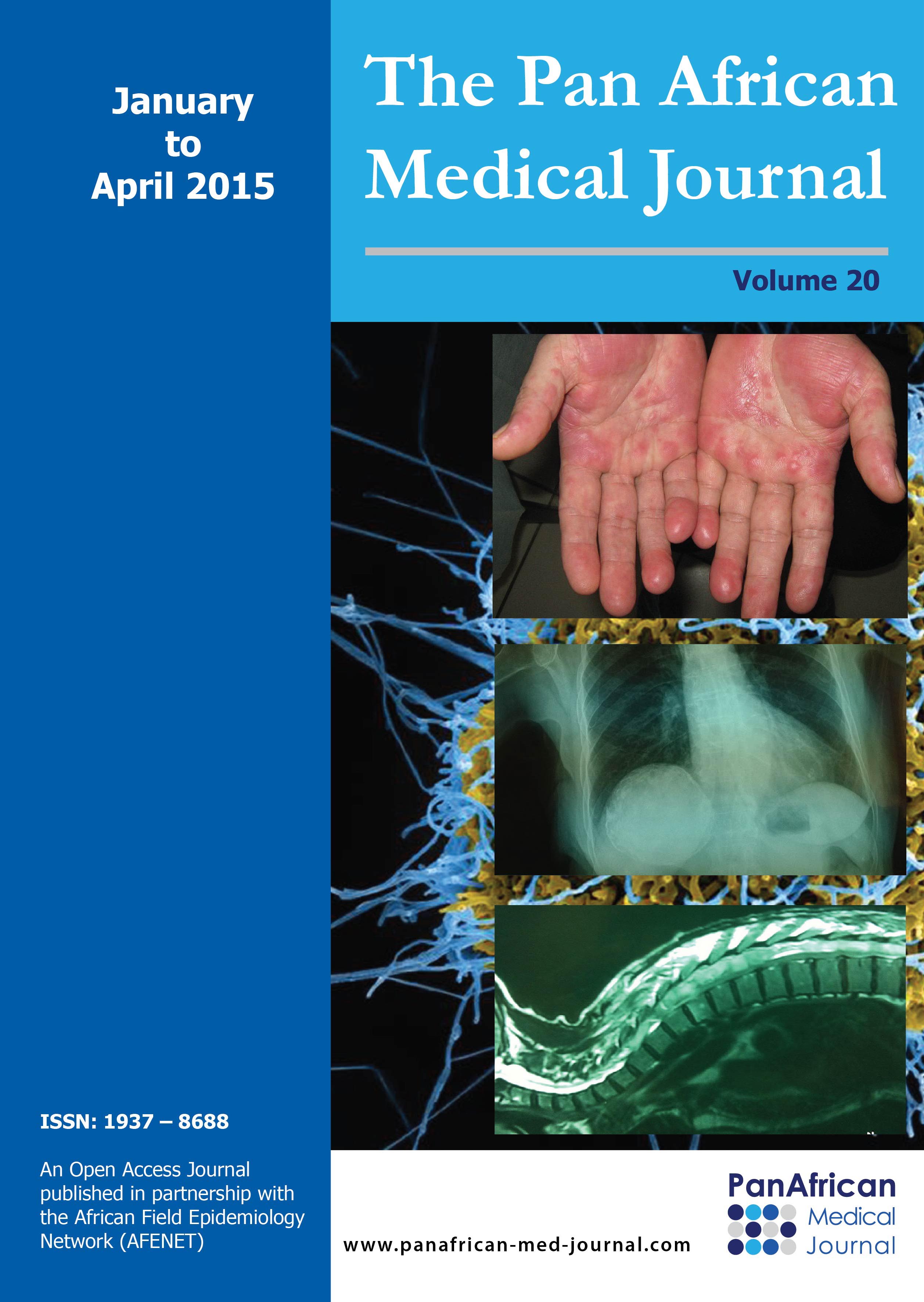
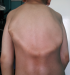
M.M, jeune garçon de 10 ans sans antécédents familiaux particuliers, consultait pour ankylose des deux membres supérieurs ŕ l'origine d'une impotence fonctionnelle invalidante. L'histoire de la maladie remontait ŕ l'âge de quatre ans, par l'apparition spontanée, de façon bilatérale et asymétrique de tuméfactions douloureuses au niveau des muscles paravertébraux, augmentant progressivement de volume pour se fusionner et s'étendre jusqu'au niveau de l'extrémité supérieure des deux diaphyses humérales, limitant considérablement la mobilité de ces derniers et gęnant sa vie quotidienne. L'évolution était marquée par l'apparition d'autres nodosités osseuses touchant les muscles parastérnales droit, ainsi que l'installation d'une cyphose dorsale d'aggravation progressive. A l'examen, il avait un bon développement psychomoteur et un discret retard staturo-pondéral. Il présentait une attitude guindé et une posture penché vers l'avant du fait de la cyphose dorsale et les nodules sous cutanés étaient immobiles, indolores, de consistance dure évoquant des excroissances osseuses, s'étendant en bande du gril costal postérieur ŕ la face postéro-médiale des extrémités supérieures des deux bras latéralement et le long de rachis dorsale en haut plus marqué du coté gauche. Ces nodules étaient également présents au niveau parasternal droit (Figure 1). Les articulations scapulo-humérales avaient un secteur de mobilité nul dans toutes les directions. Les articulations des coudes, genoux et hanches étaient libres. L'examen des pieds a trouvé un hallux valgus bilatéral. Le bilan phosphocalcique était normal. La radiographie thoracique de face et de profil objectivait une ossification irréguličre s'étendant en bande de rachis jusqu'au niveau des deux omoplates et des deux humérus, sans lésions parenchymateuses pulmonaires (Figure 2). Le scanner thoracique avec reconstruction montrait la présence d'une importante ossification extrasquelletique engainant la face postérieure du tronc, et fusionnant avec les extrémités supérieures des humérus, associée ŕ d'autres ossifications corticalisées en parasternale et en paravertébrale droit, et ŕ une cyphose dorsale sans lésions parenchymateuses pulmonaires (Figure 3). La scintigraphie osseuse a montré plusieurs foyers d'hyperfixation (Figure 4). L'aspect de ces ossifications et leur association ŕ un hallux valgus, nous ont permis de poser le diagnostic de myosite ossifiante progressive. Peur de favoriser leur extension, aucune biopsie ou intervention de libération de ces ossifications hétérotopiques n'a été indiquée.
La MOP, affection génétique extręmement rare, touche en moyenne 1 personne sur 2 millions. Décrite pour la premičre fois en 1648 par PA TIN [1]. Cette affection est décrite essentiellement chez le jeune enfant et touche indifféremment les deux sexes: 41% des MOP sont dépistés avant l'âge de 2ans, 80% avant 10ans et 95% avant 15 ans [2]. Notre cas est en harmonie avec la littérature, les premiers symptômes s'étant manifestés ŕ l'âge de 4 ans. Il ne semble pas que l'ethnie, la race, le sexe ou la géographie constituent des facteurs de prédisposition, cependant un facteur génétique est vraisemblable avec la survenue d'une mutation spontanée du gčne ACVR1 portée sur le chromosome 4 de transmission autosomique dominante, codant pour un récepteur des protéines BMP4 impliquées dans la croissance et le modelage de l'os [3]. La symptomatologie de la MOP associe une fébricule ŕ l'apparition de tuméfactions inflammatoires touchant les tissus conjonctifs et les muscles. Celles-ci vont s'ossifier et s'étendre progressivement ŕ toutes les régions du corps. Ce processus affecte initialement les muscles du cou et les muscles paravertébraux avec une extension suivant un schéma proximo-distal et crânio-caudal [4]. Les atteintes vont alors toucher les épaules puis le rachis lombaire puis les hanches. Plus tard les articulations distales peuvent ętre atteintes vers la 3čme décennie de la vie. Cependant selon Munchmeyer, les muscles qui ne s'insčrent pas sur le squelette par leurs deux extrémités sont épargnés. Ce qui explique le respect des muscles oculaires, du diaphragme, de la langue, du pharynx, du larynx et des muscles lisses [4].
Notre cas présente une grande similitude avec la littérature concernant les muscles cibles et la progression des lésions et tend ŕ confirmer l'hypothčse de Munchmeyer. Par ailleurs, des malformations osseuses essentiellement présentes au niveau des pouces et des gros orteils sont mises en évidence dans 70ŕ 100% des cas [4]. Elles se traduisent cliniquement par une microdactylie et un hallux valgus bilatéral. D'autres malformations plus rares et trčs variées sont décrites telles que des anomalies vertébrales, épiphysaires, rotuliennes, des épines calcanéennes, du col fémoral, des cinquičmes doigts ou encore des épaississements de la corticale interne du tibia. Dans notre cas, seul un hallux valgus bilatéral était observé. Des patients atteints de formes atypiques de FOP ont été décrits. Soit les signes classiques de la FOP sont présents, avec un ou plusieurs signes atypiques (par exemple: anémie aplastique intercurrente, craniopharyngiome, glaucome infantile ou retard de croissance) (FOP "plus"), soit l'un ou les deux signes cardinaux de la FOP présentent des variations majeures par exemple, des gros orteils normaux ou une réduction sévčre des doigts (variantes de la FOP). En raison de l'ankylose des articulations costo-vertébrales et la déformation vertébrale, les patients finissent par développer une insuffisance respiratoire restrictive avec une atélectasie. La pneumonie et l'insuffisance cardiaque droite peuvent ętre mortelles. Les examens biologiques font partie des éléments diagnostiques de la MOP mais ils ont peu d'intéręt thérapeutique, évolutif et pronostic [5]. Le minimum d'examens biologiques ŕ demander est relatif au métabolisme osseux dont le bilan phosphocalcique et le taux de PAL et celui relatif au processus inflammatoire dont la VS et la numération formule sanguine [5]. La radiologie conventionelle est la clé du diagnostic en montrant des images évocatrices telles qu'une calcification corticalisée ectopiques des muscles atteints et ŕ un stade évolué, des ponts osseux entre les différentes parties du squelette avec un véritable squelette ectopique. Elle permet aussi de montrer des malformations osseuses congénitales typiques. Les autres moyens d'imagerie ne sont pas nécessaires pour le diagnostic, surtout ŕ un stade évolué. Le scanner permet de mieux analyser les ossifications ainsi que leur étendue grâce aux reconstructions multiplannaires tels observés chez notre malade. L'IRM et la scintigraphie peuvent montrer des lésions au début non encore ossifiées [6]. Le diagnostic de la FOP est radio-clinique ne nécessitant pas de biopsie qui peut ętre le point de départ d'une ossification ectopique et peut induire en erreur, vu le caractčre hétérogčne des lésions [6].
Plusieurs diagnostics différentiels peuvent ętre évoqués surtout au début de la maladie et lorsqu'on n'examine pas les orteils et les doigts. Les métastases ossifiantes, les hématomes ossifiés, les tendinopathies calcifiantes, les maladies exostosantes, la myosite ossifiante circonscrite post-traumatique qui est généralement limitée ŕ une seule localisation, délimitée, et douloureuse, qui survient ŕ la suite de traumatismes. Les sarcomes des tissus mous peuvent ętre évoqués sur les résultats de la biopsie des lésions précoces [7, 8]. L'ostéodystrophie héréditaire d'ALBRIGHT est caractérisée par une ossification dans le muscle et le tissu conjonctif, mais plus marquée dans la graisse sous-cutanée et associée ŕ une pseudohypoparathyroidie [9]. La spondylarthrite ankylosante pourra ętre évoquée devant une ankylose du rachis et des articulations sacro-iliaques [10]. L'histoire naturelle de cette affection est caractérisée par des poussées inflammatoires de 2 ŕ 3 semaines, entrecoupées par des périodes de latence plus ou moins longues. L'évolution peut ętre émaillée par la survenue de plusieurs complications thromboemboliques, neurologiques, cutanées, infectieuses ou respiratoires. Son pronostic dépend surtout de la survenue d'une insuffisance respiratoire ŕ un stade tardif.
Ŕ ce jour, aucun traitement n'a prouvé son efficacité. Cependant plusieurs moyens thérapeutiques sont proposés dans la prise en charge des MOP [1]. La kinésithérapie douce est proposée ŕ visée antalgique lors des poussées. Le traitement médicamenteux fait appel aux corticoďdes, aux inhibiteurs des mastocytes, aux inhibiteurs de la cyclo-oxygénase 2, aux anti-inflammatoires non stéroďdiens aux amino-biphosphonates, aux antagonistes de BMP, aux agents anti-angiogéniques ou encore aux rétinoďdes avec des résultats encore aléatoires et insatisfaisants. Leur utilisation doit ętre mise en balance avec la sévérité potentielle de leurs effets secondaires. Il y'a peu de place pour la chirurgie d'autant plus que l'anesthésie de ces patients est difficile en raison de la rigidité rachidienne et de la fixation des mandibules [11, 12]. L'ablation chirurgicale de ces ossifications afin de mobiliser les articulations constitue un nouveau traumatisme qui favorise le développement d'ossifications hétérotopiques supplémentaires. Le seul but de la chirurgie est de corriger les attitudes vicieuses pour donner un enraidissement en une position la plus favorable possible. Devant les limites des traitements médical et chirurgical, la prévention devient trčs importante. Plusieurs mesures peuvent limiter l'évolution de la maladie: prévention des traumatismes, du blocage mandibulaire, médecine de réhabilitation et physiothérapie avec kinésithérapie respiratoire [13].
Maladie extręmement rare, la MOP doit ętre évoquée chez tout enfant présentant une ossification hétérotopique des parties molles, associée ŕ des anomalies osseuses congénitales. Son diagnostic est radio-clinique. L'évolution est marquée par l'apparition de raideurs articulaires particuličrement invalidantes. Elle peut aussi mettre en jeu le pronostic vital de par les complications d'ordre général qu'elle occasionne. Aucun traitement préventif ou curatif efficace n'est disponible ŕ ce jour. Néanmoins, plusieurs molécules sont en cours d'expérimentation
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1: aspect clinique du tronc montrant une cyphose dorsale, une raideur de tous le tronc, avec coulées osseuses sous cutanées, en arceau, en regard de grille costale postérieure reliant les deux humérus
Figure 2: radiographie thoracique profil: calcifications corticalisées irréguličres s’étendant en bande de rachis jusqu’au niveau des deux omoplates et des deux humérus
Figure 3: TDM thoracique avec reconstruction: importante ossification extrasquelletique engainant la face postérieure du tronc, dessinant un arceau a concavité supérieure et fusionnant avec les extrémités supérieures des humérus, associée ŕ d’autres ossifications corticalisées en parasternale et en paravertébrale droit, et ŕ une cyphose dorsale
Figure 4: scintigraphie osseuse a montré plusieurs foyers d’hyperfixation
- Kaplan FS, Shore EM, Connor JM. The International Clinical Consortium on FOP: The medical management of fibrodysplasia ossificans progressive: current treatment considerations. Clin Proc Int Consort FOP. 2008; 3: 1-82. PubMed | Google Scholar
- Cohen RB, Hahn GV, Tabas JA, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva: a study of forty-four patients. J Bone Joint Surg Am. 1993; 75(2): 215-9. PubMed | Google Scholar
- Groppe JC, Shore EM, Kaplan FS. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin Orthop Relat Res. 2007 Sep; 462: 87-92. PubMed | Google Scholar
- Connor JM, Evans DA. Fibrodysplasia ossificans progressiva: the clinical features and natural history of 34 patients. J Bone Joint Surg Br. 1982; 64 (1): 76-83. PubMed | Google Scholar
- Bhatia M, Bonnet D, Wu D, et al. Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. J Exp Med. 1999 Apr; 189(7): 1139-48. PubMed | Google Scholar
- Bridges AJ, Hsu KC, Singh A, Churchill R, Miles J. Fibrodysplasia (myositis) ossificans progressiva. Semin Arthritis Rheum. 1994 Dec; 24(3): 155-64. PubMed | Google Scholar
- Liu K, Tripp S, Layfield LJ. Heterotopic ossification: review of histologic findings and tissue distribution in a 10-year experience. Path Res Pract. 2007; 203(9): 633-40. PubMed | Google Scholar
- Ragunanthan N, Sugavanam C. Pseudomalignant myositis ossificans mimicking osteosarcoma: a case report. J Orthop Surg (Hong Kong). 2006 Aug; 14(2): 219-21. PubMed | Google Scholar
- Athanasou NA, Benson MK, Brenton BP, Smith R. Progressive osseous heteroplasia: a case report. Bone. 1994 Sep-Oct; 15(5): 471-5. PubMed | Google Scholar
- Elloumi M, Fourati H, Ezeddine M, Baklouti S. Myositis ossificans progressiva mimicking ankylosing spondylitis (a case report). Joint Bone Spine. 2006 Oct; 73(5): 570-1. PubMed | Google Scholar
- Singh A, Ayyalapu A, Keochekian A. Anesthetic Management in Fibrodysplasia Ossificans Progressiva (FOP): a case report. J Clin Anesth. 2003 May; 15(3):211-3. PubMed | Google Scholar
- Tumolo M, Moscatelli A, Silvestri G. Anaesthetic management of a child with fibrodysplasia ossificans progressiva. Br J Anaesth. 2006 Nov; 97(5): 701-3. PubMed | Google Scholar
- Levy C, Berner TF, Sandhu PS, McCarty B, Denniston NL. Mobility Challenges and Solutions for Fibrodysplasia ossificans progressiva. Arch Phys Med Rehabil. 1999 Oct; 80(10): 1349-53. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ








Authors´ services
