
Syndrome de Sweet: ťtude clinique et natomopathologique sur 5 ans
Hanae Bouzidi, Salim Gallouj, Nissrine Amraoui, Fatima Zahra Mernissi, Taoufiq Harmouch
Corresponding author: Hanae Bouzidi, 10 rťsidence Bial Nį2, Bouevard Medina Monawara, Quartier Amal, Narjiss, FŤs 30070, Maroc 
Received: 30 Mar 2014 - Accepted: 18 Jan 2015 - Published: 14 Apr 2015
Domain: Clinical medicine
Keywords: Syndrome de Sweet (SS), dermatose aiguë fébrile, lésions de vascularite
©Hanae Bouzidi et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Hanae Bouzidi et al. Syndrome de Sweet: ťtude clinique et natomopathologique sur 5 ans. Pan African Medical Journal. 2015;20:362. [doi: 10.11604/pamj.2015.20.362.4274]
Available online at: https://www.panafrican-med-journal.com//content/article/20/362/full
Original article 
Syndrome de Sweet: ťtude clinique et natomopathologique sur 5 ans
Syndrome de Sweet: ťtude clinique et natomopathologique sur 5 ans
Hanae Bouzidi1,&, Salim Gallouj1, Nissrine Amraoui1, Fatima Zahra Mernissi1, Taoufiq Harmouch2
1Service de Dermatologie-Vťnťrologie, CHU Hassan II, FŤs, Maroc, 2Service díAnatomopathologie, CHU Hassan II, FŤs, Maroc
&Auteur correspondant
Hanae Bouzidi, 10 rťsidence Bial Nį2, Bouevard Medina Monawara, Quartier Amal, Narjiss, FŤs 30070, Maroc
Le syndrome de Sweet ou dermatose aiguŽ fťbrile neutrophilique est une maladie inflammatoire rare ŗ expression cutanťe prťdominante, appartenant au groupe des dermatoses neutrophiliques. Il est caractťrisť par le polymorphisme de son expression clinique et la diversitť des maladies qui peuvent lui Ítre associťes. Le but de ce travail ťtait d'ťtudier les particularitťs cliniques, anatomopathologiques, thťrapeutiques et ťvolutives du syndrome de Sweet. Nous rapportons une ťtude rťtrospective et descriptive de 25 cas de syndrome de Sweet observťs dans les services de dermatologie et d'anatomie pathologique du centre hospitalier universitaire de FŤs sur une pťriode de 5 ans. Notre sťrie ťtait constituťe de 5 hommes et de 20 femmes avec un sex-ratio (hommes/femmes) de 0,25. L'‚ge moyen des patients ťtait de 47 ans avec des extrÍmes allant de 11 ŗ 75 ans. Des maladies associťes ťtaient retrouvťes chez 17 patients : hťmopathies (deux cas), tumeur solide (1cas), maladie chronique de l'intestin (1cas), tuberculose (1 cas), diabŤte (trois cas) et infections (9 cas). Deux patientes ťtaient enceintes au moment du diagnostic. Les manifestations cutanťes ťtaient polymorphes avec atteintes muqueuses dans deux cas. Les lťsions siťgeaient le plus souvent au niveau acral. Histologiquement, le derme ťtait le siŤge d'un infiltrat dense et diffus riche en polynuclťaires neutrophiles dans tous les cas. Une inflammation de la paroi des vaisseaux ťtaient observťes dans trois cas. Le syndrome de Sweet peut Ítre rťvťlateur ou prťcťder des maladies associťes, ce qui impose une surveillance rigoureuse et prolongťe.
Le syndrome de Sweet (SS), ou dermatose aiguŽ fťbrile neutrophilique, a ťtť dťcrit pour la premiŤre fois par Robert Douglas Sweet en 1964 [1]. Il appartient au groupe des dermatoses neutrophiliques. Il peut Ítre considťrť comme la plus typique des entitťs de ce groupe [2-4]. Il est caractťrisť par le polymorphisme de son expression clinique et la diversitť des maladies qui peuvent lui Ítre associťes. Il se manifeste classiquement par une fiŤvre, une ťruption de plaques ou de nodules ťrythťmateux cutanťs, une hyperleucocytose ŗ polynuclťaires neutrophiles (PNN) et un infiltrat dermique massif constituť essentiellement de PNN. Le traitement repose sur des traitements anti-inflammatoires qui permettent une rťsolution rapide des symptŰmes et des lťsions cutanťomuqueuses [2]. Nous ťtudierons les caractťristiques cliniques, anatomopathologiques, thťrapeutiques et ťvolutives de cette affection ŗ partir d'une ťtude rťtrospective et descriptive d'une sťrie de 25 cas de SS.
Les dossiers de 25 cas de SS diagnostiquťs dans notre institution du 1er janvier 2009 au 1er janvier 2014 ont ťtť revus. Une fiche analytique complŤte a ťtť ťtablie pour chaque patient. Tous les patients ont eu un examen clinique complet, une numťration de la formule sanguine, une vitesse de sťdimentation, un dosage de la protťine C-rťactive (CRP), et une biopsie cutanťe. Le diagnostic positif de SS ťtait ťtabli sur les critŤres cliniques, biologiques et histologiques (Tableau 1). Le diagnostic a ťtť retenu devant la prťsence de deux critŤres majeurs et deux critŤres mineurs. Le syndrome d'immunodťficience acquise a ťtť recherchť chez tous nos patients par une sťrologie HIV, les hťmopathies par une EEP, Les autres maladies associťes ont ťtť recherchťes par des explorations biologiques et radiologiques en fonction des signes d'appel. Nous avons procťdť au cours de cette ťtude ŗ une analyse dťtaillťe de notre sťrie en fonction de l'‚ge, du sexe, des antťcťdents, de la prťsentation clinique, des donnťes biologiques, de l'aspect anatomopathologique, du traitement et des diffťrentes modalitťs ťvolutives.
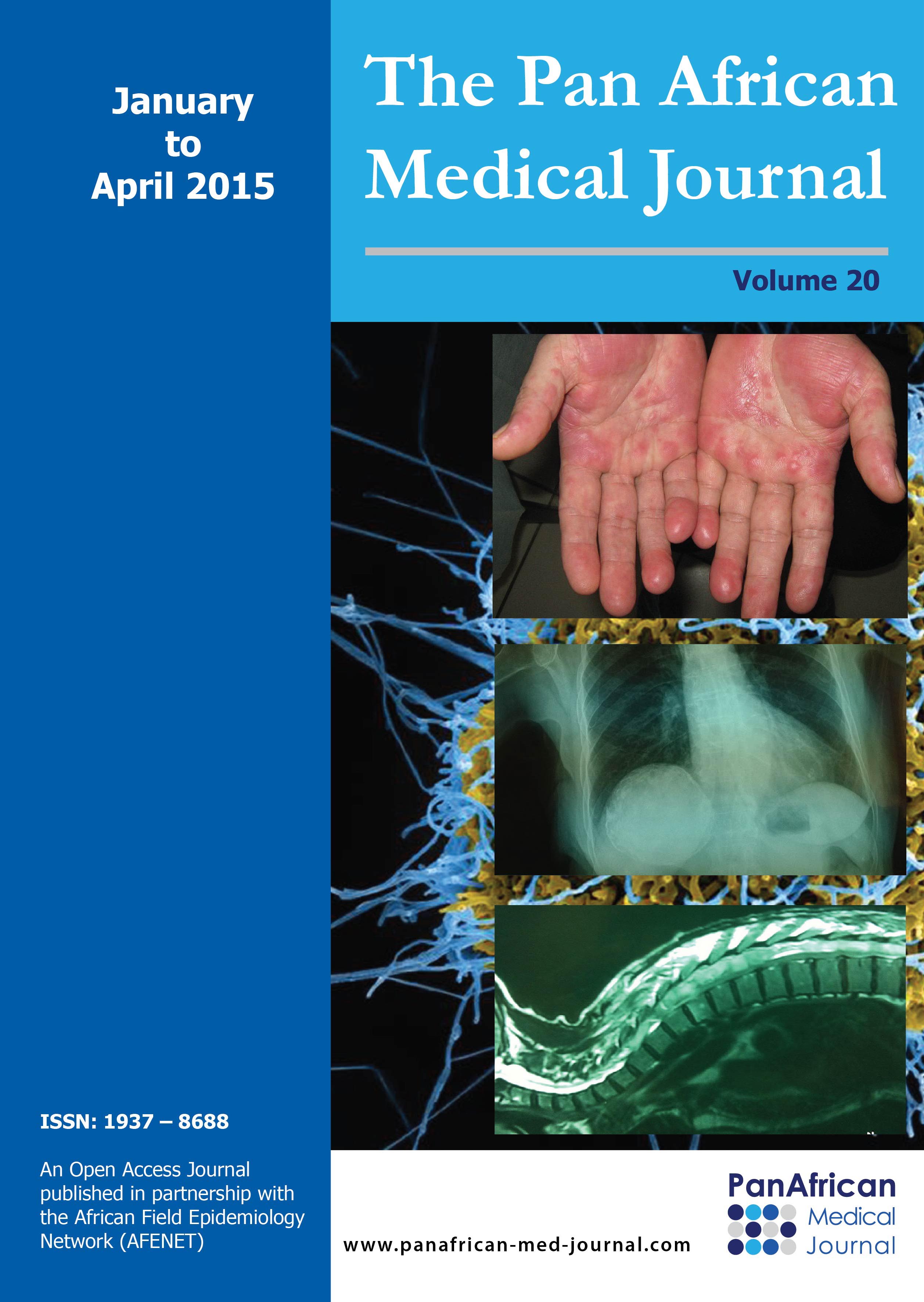
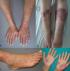
Entre le mois de janvier 2009 et le mois de janvier 2014, nous avons colligť 45 cas de dermatoses neutrophiliques qui se rťpartissaient comme suit: 25 cas de SS (55,5%), 15 cas de pyoderma gangrenosum (33,3%), 3 cas de pustulose sous-cornťe (6,6%), 2 cas d'erythema elevatum diutinum (4,4%). Notre sťrie de SS ťtait constituťe de 5 hommes et 20 femmes avec un sex-ratio (homme/femme) de 0,25. L'‚ge moyen des patients ťtait de 47 ans avec des extrÍmes allant de 11 ŗ 75 ans et un pic entre la quatriŤme et la cinquiŤme dťcennie. Des maladies associťes ťtaient retrouvťes chez 17 patients. Il s'agissait d'infections dans 9 cas (angines, ťrysipŤle, cellulite frontale et maxillaire, sinusite et dacryocystite), une hťmopathie dans 2 cas (leucťmie lymphoÔde chronique et leucťmie myťloÔde aigue), une tumeur solide dans un cas (adťnocarcinome mammaire), une maladie cœliaque dans un cas, une tuberculose dans un cas et un diabŤte dans trois cas. Deux cas ťtaient associťs ŗ une prise mťdicamenteuse (cycline et quinolone). Par ailleurs, deux femmes ťtaient enceintes, ŗ 16 et 28 semaines d'amťnorrhťe. La symptomatologie ťtait d'installation brutale dans tous les cas. Le dťlai entre le dťbut des premiers symptŰmes et la premiŤre consultation variait entre un jour et trois mois. ņ la phase prodromique, une symptomatologie fonctionnelle ťtait rapportťe dans 21 cas (84%). Il s'agissait d'une fiŤvre (19 cas), d'un syndrome pseudogrippal (sept cas), d'arthralgies (10 cas), d'une conjonctivite (5 cas), ou d'une toux sŤche (un cas). ņ la phase d'ťtat, une ťruption cutanťe ťtait prťsente chez tous les patients. Elle consistait en des plaques papuleuses ťrythťmateuses ou violines infiltrťes bien limitťes, de taille variable, dans 19 cas (Figure 1). Des aspects atypiques ťtaient prťsents dans 18 cas: des lťsions en cocarde ou en pseudo-cocarde (7cas), des bulles (4 cas), des pustules (4 cas) ou des vťsicules (deux cas) (Figure 2). Ces lťsions ťtaient retrouvťes sur les membres supťrieurs dans 15 cas, sur les membres infťrieurs dans 11 cas, dans la rťgion de la tÍte et du cou dans 19 cas et du tronc dans deux cas. 2 atteintes muqueuses ťtaient notťes dans notre sťrie. La rťpartition des manifestations extra-cutanťes chez les patients ťtait par ordre de frťquence dťcroissant: osteoraticulaire dans 14% des cas, oculaire 10%, rťnal dans 7% des cas, neurologique, musculaire et pulmonaire dans 3% des cas chacun.
†
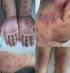
La numťration formule sanguine montrait une hyperleucocytose dans 16 cas (64 %). Cette hyperleucocytose ťtait ŗ prťdominance de polynuclťaires (PNN) (supťrieure ŗ 70%). La vitesse de sťdimentation ŗ la premiŤre heure ťtait augmentťe dans tous les cas avec une moyenne de 85,1 mm/1 h et des extrÍmes allant de 10 ŗ 140 mm/1 h. La valeur moyenne de la CRP ťtait de 57,5 mg/L (6-194). Sur le plan anatomopathologique, l'ťpiderme ťtait normal dans 13 cas (52%). Des modifications ťpidermiques ťtaient retrouvťes dans 12 cas (48 %). Elles consistaient en une hyperkťratose orthokťratosique (20%), une acanthose (12%), une pustule spongiforme (16%) ou une spongiose (4%). Le derme rťticulaire ťtait le siŤge d'une inflammation dense dans tous les cas. L'infiltrat ťtait constituť de PNN dans tous les cas, mais associait ťgalement des polynuclťaires ťosinophiles dans 5 cas (20%), des ťlťments lymphoplasmocytaires dans 10 cas (40%) et des histiocytes dans 3 cas (12%). Des dťbris nuclťaires de PNN ťtaient notťs dans 5 cas (20%). Au niveau du derme papillaire, un œdŤme ťtait retrouvť dans 12 cas (48%) et une vasodilatation dans 11 cas (44%). Une vascularite leucocytoclasique ťtait prťsente dans 3 cas (12%) (Figure 3). L'hypoderme, prťlevť dans la majoritť des cas, ťtait d'aspect normal (Tableau 2).†
L'aspect histologique ťtait typique d'un SS dans la grande majoritť des cas. Les problŤmes de diagnostic diffťrentiel avec d'autres dermatoses neurophiliques, notamment le pyoderma gangrenosum et les dermatoses infectieuses, ťtaient facilement rťsolues par la confrontation de l'aspect histologique et des donnťes clinicobiologiques. Les patients ont ťtť traitťs par des anti-inflammatoires non stťroidiens en particulier par de l'indomťtacine ŗ la dose de 150 mg/J la premiŤre semaine et 100 mg/J les deux semaines suivantes. Dans la majoritť des cas la rťponse ťtait favorable avec rťmission en quelques jours, dans les rares cas rťsistants ou rťpondant d'une maniŤre incomplŤte on a eu recours au corticoÔdes dans le cas du Sweet profond avec atteinte neurologique et ŗ la colchicine dans un autre cas de Sweet typique avec atteinte articulaire. Trois patients n'ont pas reÁu de traitement. Les patients ťtaient suivis sur des pťriodes variables allant de 3 mois ŗ 4 ans. Le suivi reposait sur un interrogatoire, un examen physique et une numťration formule sanguine. L'ťvolution ťtait favorable chez 92% des patients avec rťsolution des signes gťnťraux et locaux. Des rťcidives locales ťtaient rapportťes dans 8% des cas. Quatre patients ťtaient perdus de vue. Parmi les patients qui n'ont pas reÁu de traitement, l'ťvolution spontanťe ťtait favorable dans deux cas; le troisiŤme ťtait perdu de vue. Au cours de l'ťvolution, il n'y a pas eu de localisation neutrophilique extracutanťe observťe.
Le SS appartient au spectre des dermatoses neutrophiliques dont les plus frťquentes sont le pyoderma gangrenosum, la dermatose pustuleuse sous-cornťe, l'erythema elevatum diutinum [3]. Le SS est la forme la plus frťquente des dermatoses neutrophiliques [4]. Il est dťfini par l'association de deux critŤres majeurs et de deux critŤres mineurs selon la classification de Su et Liu, modifiťe par Von den Driesch [5]. Son incidence annuelle est ťvaluťe ŗ trois par million d'individus. Plus frťquent chez la femme, il prťdomine ŗ l'‚ge moyen de la vie mais peut survenir ŗ tout ‚ge [6]. Il survient rarement chez l'enfant (8%), sans prťdominance de sexe [7]. La pathogťnie du SS est multifactorielle [6]. L'association ŗ des maladies infectieuses, des maladies auto-immunes, des nťoplasies ou des mťdicaments plaide en faveur d'une rťaction d'hypersensibilitť [7]. Elle ferait intervenir des cytokines, l'interfťron gamma et le TNF pour l'activation et le recrutement des neutrophiles [8]. Certains auteurs insistent sur le rŰle de l'interleukine-1 (IL-1) et montrent l'efficacitť du traitement par l'antagoniste du rťcepteur de l'IL-1 dans les cas rťfractaires [9]. Cliniquement, le SS dťbute brutalement, parfois annoncť par des prodromes non spťcifiques, respiratoires ou digestifs, avec une fiŤvre, des myalgies, des arthralgies, des douleurs abdominales ou une conjonctivite [4]. ņ la phase d'ťtat, la fiŤvre est accompagnťe d'une nette altťration de l'ťtat gťnťral. L'ťruption est faite de plaques et de papules ťrythťmateuses infiltrťes, surťlevťes, bien limitťes, ŗ surface bosselťe, de siŤge dermique et hypodermique. Ces plaques ne sont pas prurigineuses, mais sensibles et douloureuses. Il peut s'agir d'une lťsion unique ou de lťsions multiples, asymťtriques. Elles prťdominent au visage, au cou, aux membres supťrieurs, mais peuvent atteindre tout le tťgument [4,6]. Les lťsions ulcťrťes survenant au niveau des muqueuses sont souvent rapportťes dans les formes associťes ŗ des hťmopathies [7]. Ces lťsions cutanťes posent un problŤme de diagnostic diffťrentiel notamment avec les maladies infectieuses, certaines vascularites ou maladies systťmiques [7].
†
Les anomalies biologiques se traduisent par une ťlťvation de la vitesse de sťdimentation supťrieure ŗ 20 mm ŗ la premiŤre heure, une ťlťvation de la CRP, une hyperleucocytose ŗ PNN supťrieure ŗ 70% [6]. Dans notre sťrie, une hyperleucocytose avec polynuclťose est notťe dans 64%. Cependant, une anťmie, une leucopťnie et une thrombocytťmie sont possibles, notamment dans les formes associťes ŗ des hťmopathies [7]. La biopsie cutanťe est nťcessaire ŗ la confirmation du diagnostic. Histologiquement, l'ťpiderme peut Ítre normal, acanthosique ou Ítre le siŤge d'une exocytose de PNN, parfois si intense qu'elle dťtermine la formation de pustules sous-cornťes uniloculaires. Il peut exister un dťcollement sous-ťpidermique rťsultant d'un 'dŤme massif [7]. Dans notre sťrie, un œdŤme est notť dans 48 % des cas. Au niveau du derme superficiel, il existe un oedŤme d'intensitť variable. L'infiltrat se dispose en une large bande avec une tendance ŗ la formation de nodules plus ou moins coalescents ŗ renforcement pťrivasculaire [10]. Il peut s'ťtendre dans le derme profond, formant des amas plus ou moins denses de topographie pťrivasculaire et pťri-annexielle. Cet infiltrat est majoritairement formť de PNN. Nťanmoins, sa composition dťpend du stade ťvolutif des lťsions : un contingent de cellules lymphocytaires peut apparaÓtre dans les stades dťbutants en particulier dans les formes associťes ŗ un syndrome myťlodysplasique [11]. Actuellement, la prťsence d'une vascularite n'est plus un critŤre d'ťlimination du diagnostic [3]. En effet, une nťcrose fibrinoÔde de la paroi des vaisseaux et une leucocytoclasie peuvent Ítre observťes dans des formes typiques de SS. Dans une sťrie de 37 cas de SS colligťs par Jordaan, une vascularite leucocytoclasique et nťcrosante est notťe dans 18 % des cas [12]. Malone et al, dans leur sťrie de 21 cas, montrent que la vascularite n'est pas un processus immunitaire primitif, mais survient secondairement suite ŗ la libťration des mťtabolites toxiques libťrťs par des neutrophiles activťs dans des lťsions anciennes [13]. Il s'agit donc d'un signe tťmoignant de l'anciennetť des lťsions. Dans notre sťrie, une vascularite leucocytoclasique est notťe dans 3 cas (12%). Au niveau de l'hypoderme, l'atteinte peut Ítre septale, lobulaire ou mixte [6]. Ces formes hypodermiques des SS semblent plus frťquentes dans les SS associťs ŗ des hťmopathies malignes, notamment aux syndromes myťlodysplasiques. Le SS pose un problŤme de classement dans le spectre des dermatoses neutrophiliques qui constituent un continuum, avec des entitťs qui sont parfois associťes [14] et s'accompagnent parfois de localisation extracutanťes comme au cours du syndrome des abcŤs aseptiques [15]. Par ailleurs, une lťsion infectieuse doit Ítre ťliminťe telle que la cellulite, l'ťrysipŤle, la tuberculose. Un prťlŤvement bactťriologique peut s'avťrer indispensable. Une fois le diagnostic posť, le problŤme essentiel sera la recherche, d'une part, de manifestations extracutanťes, d'autre part, d'une maladie associťe, essentiellement une hťmopathie [3].†
On peut distinguer plusieurs formes de SS. Les formes classiques ou idiopathiques sont les plus frťquentes (75%) [16]. Elles surviennent souvent chez la femme de la quarantaine et comportent presque toujours une polynuclťose sanguine [3]. Le SS est paranťoplasique dans 10 ŗ 20% des cas, accompagnant ou rťvťlant une affection maligne qui est hťmatologique dans 85% des cas et une tumeur solide dans 15% des cas. La leucťmie myťloÔde aiguŽ est l'hťmopathie la plus frťquemment diagnostiquťe (85% des cas). Les tumeurs solides sont le plus souvent gťnito-urinaires, mammaires ou digestives [7]. Les SS para-inflammatoires sont associťs ŗ une maladie du groupe dysimmunitaire telle que la rectocolite ulcťro-hťmorragique et la maladie de Crohn. Les infections les plus souvent rapportťes sont celles des voies respiratoires supťrieures. Les formes secondaires ŗ une prise mťdicamenteuse incriminent plusieurs classes thťrapeutiques et notamment les facteurs de croissance hťmatopoÔťtique [3]. Chez l'enfant et l'adolescent, la forme accompagnant une affection inflammatoire (33%) est la plus frťquente, suivie par la forme paranťoplasique (25 %) [17]. Dans notre sťrie, le SS ťtait idiopathique dans 10 cas (40 %) et parainflammatoire dans 10 autres cas (21,3%). Trois formes paranťoplasiques ont ťtť notťes et deux cas post mťdicamenteux. Le suivi ťtait de durťe variable entre 3 mois et 4 ans. Il s'est basť sur un interrogatoire, un examen physique et des examens complťmentaires orientťs selon les signes d'appel du patient. Au cours de l'ťvolution d'un SS, tous les organes et les muqueuses peuvent Ítre le siŤge du mÍme infiltrat neutrophilique aseptique et se manifester par des symptŰmes variťs selon l'organe atteint. L'atteinte oculaire peut Ítre la premiŤre manifestation du syndrome. Il s'agit d'une conjonctivite, d'une ťpisclťrite ou une iridocyclite. L'atteinte articulaire est frťquente et trŤs polymorphe. Elle survient dans 33 ŗ 62% des cas. Il s'agit d'une arthrite aseptique qui peut Ítre aiguŽ ou chronique, mono- ou polyarticulaire, atteignant les membres supťrieurs ou infťrieurs [7]. L'atteinte neurologique ou neuro-Sweet peut se manifester par une encťphalite ou une mťningite et pose un problŤme diagnostic avec leneuro-BehÁet. Les autres manifestations extracutanťes sont rares et peuvent Ítre pulmonaire, neurologique, cardiaque, osseuse [4]. L'approche thťrapeutique dťpend de l'extension et la profondeur des lťsions, de l'ťtat gťnťral du patient et des maladies associťes. L'ťvolution spontanťe peut Ítre favorable en quelques semaines ŗ quelques mois notamment dans les formes idiopathiques. Le traitement de la maladie associťe, quand elle est curable, peut influencer favorablement le pronostic. La corticothťrapie systťmique ŗ la dose de 0.5 ŗ 1mg/kG/J avec dťcroissance lente et progressive en quelques semaines est le traitement de choix. La corticothťrapie intralťsionnelle et les dermocorticoÔdes sont efficaces dans les formes localisťes. D'autres alternatives thťrapeutiques peuvent Ítre proposťes en premiŤre intention telles que la colchicine, indomťthacine, iodure de potassium, dapsone, clofazimine, cyclines, ciclosporine, thalidomide, isotrťtinoÔne, anakinra, mais ces mollecules ont fait l'objet de publications que sur des cas isolťs ou de trŤs petites sťries. Trois d'entre elles semblent toutefois particuliŤrement intťressantes : l'iodure de potassium (mais 2 cas de vascularites sťvŤres ont ťtť rapportťes sous ce traitement), la colchicine ŗ la dose de 1 ŗ 1,5 mg/J et l'indomťthacine ŗ la dose de 150 mg/J la premiŤre semaine et 100 mg/J les deux semaines suivantes. Recommandťs en premiŤre intention chez les personnes ‚gťes, ou en cas de contre-indication aux corticoÔdes bien que la rapiditť d'action et le faible pourcentage de rťcidives observť avec les 2 derniŤres molťcules sont des arguments pour une prescription plus large de premiŤre intention [18].†
Les corticoÔdes reprťsentent lŗ aussi le traitement de rťfťrence, ŗ la dose de 0.5 ŗ 1mg/kG/J. Ils sont trŤs efficaces avec disparition rapide des symptomes. Les rťcidives sont cependant assez frťquentes ŗ l'arrÍt (>20%), et les formes chroniques ne sont pas rares (10% ŗ 3 ans). Un certain nombre d'alternatives ont ťtť proposťes: colchicine, indomťthacine, iodure de potassium, dapsone, clofazimine, cyclines, ciclosporine, thalidomide, isotrťtinoÔne, anakinra' mais ces mollecules ont fait l'objet de publications que sur des cas isolťs ou de trŤs petites sťries. Trois d'entre elles semblent toutefois particuliŤrement intťressantes: l'iodure de potassium (mais 2 cas de vascularites sťvŤres ont ťtť rapportťes sous ce traitement), la colchicine ŗ la dose de 1 ŗ 1,5 mg/J et l'indomťthacine ŗ la dose de 150 mg/J la premiŤre semaine et 100 mg/J les deux semaines suivantes. Recommandťs en premiŤre intention chez les personnes ‚gťes, ou en cas de contre-indication aux corticoÔdes bien que la rapiditť d'action et le faible pourcentage de rťcidives observť avec les 2 derniŤres molťcules sont des arguments pour une prescription plus large de premiŤre intention Dans notre expťrience les AINS administrťs en premiŤre intention avaient des rťsultats trŤs satisfaisant incitant ŗ prťconisť cette option thťrapeutique afin d'ťviter une corticothťrapie systťmique ťtant une thťrapie lourde avec beaucoup plus d'effets secondaires.
Les rťsultats cliniques et histologiques des 25 patients atteints de SS dans notre ťtude sont gťnťralement comparables ŗ ceux publiťs dans la littťrature, avec quelques diffťrences. Ce qu'il faut retenir c'est que Le syndrome de Sweet peut prťcťder ou rťvťler des maladies associťes, ce qui nťcessite une surveillance rigoureuse et prolongťe.
Les auteurs ne dťclarent aucun conflit d'intťrÍts.
tous les auteurs ont contribuť ŗ la redaction, lu et approuvť la version finale du manuscrit.
Tableau
1: critŤres diagnostiques du syndrome de Sweet
Tableau 2: tableau rťcapitulatif des lťsions histologiques observťes chez 25 patients avec syndrome de sweet
Figure 1: (a, b, c, d) prťsence de papules et plaques ťrythťmateuses údťmateuses au niveau acral chez des patients prťsentant un syndrome de sweet
Figure 2: (e,
f) lťsions
en cocarde crouteuses ; (g, h) lťsions vťsiculeuses et bulleuses dans le cadre
de la
maladie de sweet
Figure 3: lťsions de vascularite leucocytoclasique de la paroi des vaisseaux (HE x 40)
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356. PubMed | Google Scholar
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10(5):301-312. PubMed | Google Scholar
- Soutou B, Vignon-Pennamen D, Chosidow O. Les dermatoses neutrophiliques. Rev Med Interne. 2011;32(5):306-13. PubMed | Google Scholar
- Wallach D. Les dermatoses neutrophiliques. Rev Med Interne. 2005;26(1): 41-53. PubMed | Google Scholar
- Von den Driesch P. Sweet's syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31(4):535-56.. PubMed | Google Scholar
- Cohen PR. Sweet's syndrome - a comprehensive review of an acute febrile neutrophilic dermatosis. Orph J R Dis. 2007;26(1)-2:34. PubMed | Google Scholar
- Bonamigo RR, Razera F, Olm GS. Neutrophilic dermatoses - part I. An Bras Dermatol. 2011;86(1):11-25. PubMed | Google Scholar
- Vanbiervliet G, Anty R, Schneider S, Arab K, Rampal P, Hebuterne X. Traitement d'un syndrome de Sweet et d'un ťrythŤme noueux compliquant une maladie de Crohn par anti-TNF_ (infliximab). Gastroenterol Clin Biol. 2002;26(1):295-7. PubMed | Google Scholar
- Kluger N, Gil-Bistes D, Guillot B, Bessis D. Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret) in a case of refractory Sweet's Syndrome. Dermatology. 2011;222(2):123-7. PubMed | Google Scholar
- Nischal KC, Khopkar U. An approach to the diagnosis of neutrophilic dermatoses: a histopathological perspective. Indian J Dermatol Venereol Leprol. 2007;73(4):222-30. PubMed | Google Scholar
- Vignon-Pennamen MD, Juillard C, Rybojad M, Wallach D, Daniel MT, Morel P, et al. Chronic recurrent lymphocytic Sweet Syndrome as a predictive marker of myelodysplasia. Arch Dermatol. 2006;142(9):1170-6. PubMed | Google Scholar
- Jordaan HF. Acute febrile neutrophilic dermatosis: a histopathological study of 37 patients and a review of the literature. Am J Dermatopathol. 1989;11(2):99-111. PubMed | Google Scholar
- Malone JC, Slone SP, Wills-Frank LA, Fearneyhough PK, Lear SC, Goldsmithn LJ, et al. Vascular inflammation (vasculitis) in Sweet Syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol. 2002;138(3):345-9. PubMed | Google Scholar
- Audemard A, Verger H, Gendrot A, Jeanjean C, Auzary C, Geffray L. Association pyoderma gangrenosum, pustulose sous-cornťe et abcŤs aseptiques splťniques: ę une maladie neutrophilique Ľ. Rev Med Interne. 2012;33(1):e28-30. PubMed | Google Scholar
- Andrť M, AumaÓtre O. Le syndrome des abcŤs aseptiques. Rev Med Interne. 2011;32(11):678-88. PubMed | Google Scholar
- Van Denhove A, Freymond N, Isaac S, Marrou K, Balme B, Gormand F, et al. Syndrome de Sweet rťvťlant un carcinome bronchique ťpidermoÔde. Rev Mal Respir. 2007;24(1):77-80. PubMed | Google Scholar
- Hospach T, von den Driesch P, Dannecker GE. Acute febrile neutrophilic dermatosis (Sweet's syndrome) in childhood and adolescence: two new patients and review of the literature on associated diseases. Eur J Pediatr. 2009; 168(1): 1-9. PubMed | Google Scholar
- Cohen PR, Kurzrock R. Sweet's syndrome: a review of current treatment options. Am J Dermatol. 2002; 3(2): 117-31. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
