
Chondrosarcome myxoďde extra-osseux: ŕ propos d’un cas et revue de la littérature
Youness Sasbou, Abdelkarim Rhanim, Younes Mhammdi, Mustapha Nkaoui, Ahmed El Bardouni, Mohammed Saleh Berrada, Mourad El Yacoubi
Corresponding author: Youness Sasbou, Service de Traumatologie-Orthopédie, CHU Ibn Sina, Rabat, Maroc 
Received: 20 Feb 2015 - Accepted: 03 Apr 2015 - Published: 14 Apr 2015
Domain: Public Health
Keywords: Chondrosarcome, extra osseux, parties molles, myxoïde, membres
©Youness Sasbou et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Youness Sasbou et al. Chondrosarcome myxoďde extra-osseux: ŕ propos d’un cas et revue de la littérature. Pan African Medical Journal. 2015;20:360. [doi: 10.11604/pamj.2015.20.360.6384]
Available online at: https://www.panafrican-med-journal.com//content/article/20/360/full
Original article 
Chondrosarcome myxoďde extra-osseux: ŕ propos d’un cas et revue de la littérature
Chondrosarcome myxoďde extra-osseux: ŕ propos d’un cas et revue de la littérature
Youness Sasbou1,&, Abdelkarim Rhanim1, Younes Mhammdi1, Mustapha Nkaoui1, Ahmed El Bardouni1, Mohammed Saleh Berrada1, Morad El Yaacoubi1
1Service de Traumatologie-Orthopédie, CHU Ibn Sina, Rabat, Maroc
&Auteur correspondant
Youness Sasbou, Service de Traumatologie-Orthopédie, CHU Ibn Sina, Rabat, Maroc
Le chondrosarcome primitif des tissus mous est beaucoup plus rare que son homonyme intra-osseux, le type myxoďde est une entité distincte sur le plan clinique, histologique, immuno-histo-chimique, cytogénétique et évolutif. Le but de cette étude est d'évaluer les caractéristiques de cette tumeur et de mettre en évidence sa difficulté diagnostique et thérapeutique. Cette étude rapporte un cas de chondrosarcome myxoďde extra-osseux atteignant les parties molles, il s'agit d'une femme âgée de 28 ans ayant une localisation au niveau de la face antéro-externe du tiers moyen de la cuisse gauche, la patiente a été explorée par une IRM. L'examen histologique a confirmé le diagnostic et la patiente a bénéficié d'une résection large complétée par une chimiothérapie adjuvante, et le suivi n'a déploré aucune récidive ni métastase. Le diagnostic des chondrosarcomes primitifs extra osseux est particuličrement histologique et leur traitement repose sur la résection chirurgicale large suivie d'une chimiothérapie adjuvante.
Le chondrosarcome myxoďde extra-squelettique est une tumeur maligne rare décrite pour la premičre fois en 1953 par Stout et Verner [1]. Elle représente moins de 3% des sarcomes des tissus mous [2]. Appelée également sarcome chordoďde, elle est actuellement classée parmi les tumeurs ŕ différenciation incertaine, en raison du manque d'arguments probants en faveur d'une différenciation cartilagineuse. Son pronostic est sombre, jalonné de récidives locales et de métastases principalement pulmonaires. Cette observation nous permet de rappeler les caractéristiques de cette tumeur en insistant sur la difficulté du diagnostic surtout anatomo-pathologique.
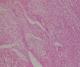
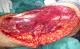
Il s'agit de Mme N.F, âgée de 28 ans et sans antécédents pathologiques particuliers, qui a consulté pour une tuméfaction sous cutanée, de la face externe du tiers supérieur de la cuisse gauche, de 10 cm de diamčtre environ, évoluant depuis 1 année et ayant augmenté de volume depuis cinq semaines environ. L'examen IRM du membre inférieur montrait une masse tumorale assez bien limitée de 8X13X14,6 cm, siégeant en sous cutanée au niveau de la face externe du tiers supérieur de la cuisse gauche (Figure 1),multi loculée, de signal hyper intense en T1, hypo intense en T2, discrčtement hétérogčne, encapsulée sans extension osseuse ni intra- musculaire. Le bilan d'extension générale était négatif. Une biopsie chirurgicale a été réalisée. L'examen histologique montrait une prolifération tumorale d'architecture nodulaire délimitée par des septas fibro-vasculaires qui abritent des cellules ŕ cytoplasme abondant éosinophile avec des noyaux ronds chromatiques, ces cellules se disposent en cordons et en pseudo acini en périphérie de ces nodules (Figure 2). Donc l'analyse histologique a conclu ŕ un chondrosarcome myxoďde extra-squelettique. La patiente a bénéficié d'une exérčse complčte de la tumeur (Figure 3), suivie d'une poly-chimiothérapie de 6 semaines. A 12 mois de recul, on ne notait pas de récidive locale ni de métastases ŕ distance.
Le CME est une entité rare distincte sur le plan clinique, histologique, immuno-histochimique, cytogénétique et évolutif. Sur le plan clinique, elle survient chez le sujet adulte vers l'âge de 50 ans avec une prédominance masculine et un sex-ratio de 2/1 [3]. Les membres représentent la localisation la plus fréquente (80 %); son sičge est en fait ubiquitaire, mais une localisation au niveau de la cuisse et du genou doit attirer l'attention [4]. Les autres sites possibles sont: les doigts, intracrânien, rétro-péritoine, plčvre et os [5]. Les signes cliniques ne sont pas spécifiques; souvent, il s'agit de douleurs ou d'une tuméfaction qui peut ulcérer la peau. Les images radiologiques ne sont pas spécifiques: un aspect pluri-nodulaire et un hyper signal en T2 sont observés ŕ l'IRM. Macroscopiquement, elle se présente sous la forme d'une masse bien limitée par une capsule fibreuse, ferme, homogčne, grise ou gélatineuse ŕ la coupe. La taille moyenne rapportée est de 7 cm (1-25 cm). L'aspect histo-pathologique usuel est celui d'une tumeur d'architecture pluri-nodulaire, faite de massifs, de lobules et/ou de travées. Les cellules tumorales sont monotones, de taille moyenne, le plus souvent ronde ou légčrement allongées, au cytoplasme clair ou éosinophile. Des inclusions rhabdoďdes sont observées dans 10 % des cas. Le noyau est en général de petite taille, réniforme ou ovoďde, ŕ chromatine fine, laissant apparaître un petit nucléole. Les atypies cyto-nucléaires et l'activité mitotique sont en général discrčtes. Dans les formes typiques, les cellules sont dispersées dans une trame plus ou moins abondante, volontiers myxoďde. Des foyers de différenciation cartilagineuse sont exceptionnellement observés. La composante cellulaire peut ętre plus ou moins dense. Des variantes plus cellulaires sont décrites, avec des dispositions cellulaires en nappes (formes solides), mais aussi des formes pseudo-épithéliales et pseudo-glandulaires. Dans les formes pauci-cellulaires, les cellules peuvent ętre fusiformes, rappelant la morphologie des myo-fibroblastes. La matrice myxoďde est colorée par le bleu Alcian aux pH 2,5 et 1, coloration résistante ŕ la hyaluronidase, suggérant la présence de sulfates chondroďtiniques. Les cellules tumorales expriment toujours la vimentine, dans environ un tiers des cas la protéine S 100 (marquage typiquement faible et focal), et souvent le CD 99. La cytokératine et l'EMA sont parfois exprimés de façon focale [1]. Les autres marqueurs ne sont pas exprimés, sauf parfois la NSE, la synaptophysine, plus rarement la chromogranine, suggérant une possible différenciation neuroendocrine [1,2]. Le CMES est associé le plus souvent ŕ une translocation chromosomique récurrente t(9;22) (q22;q12), conduisant ŕ un réarrangement génétique entre le gčne EWS, localisé sur le chromosome 22, et le gčne TEC, localisé sur le chromosome 9 [1]. Le réarrangement peut ętre recherché par RT-PCR sur tissu fixé. D'autres translocations sont plus rarement observées [2]: t(9;15) (q22; q12), t(9;17) (q22;q11.2), t(9;17;15) (q22;q11;q22), t(2;13) (q32;p12), et t(11;22) (q11;p11). ur le plan évolutif, le CMES est caractérisé par une agressivité locale par rapport ŕ leur homologue squelettique, avec des récidives locales assez fréquentes (50 %). L'évolution est lente avec une survie prolongée et le pronostic ŕ long terme est défavorable (30 ŕ 70 % de survie ŕ dix ans) [3,6]. Les métastases surviennent dans la moitié des cas, elles sont de localisations souvent pulmonaires, parfois au niveau des tissus mous, de l'os, des ganglions ou du cerveau. Ŕ l'heure actuelle, il n'y a pas de facteurs pronostiques bien établis, et ils sont pour la plupart sujet ŕ controverse. Les principaux facteurs de mauvais pronostic rapportés sont la survenue chez l'homme, l'âge tardif de survenue, une taille tumorale supérieure ŕ 10 cm, le sičge proximal, le caractčre incomplet de l'exérčse chirurgicale initiale, l'absence de réséquabilité et la découverte de métastases au moment du diagnostic [1, 6,7]. Les critčres histo-pronostiques tels que la nécrose, l'activité mitotique, le degré de différenciation ne semblent pas influencer le pronostic [8,9]. Le traitement fait appel ŕ la chirurgie premičre de la tumeur initiale et/ou des métastases, la radiothérapie et la chimiothérapie en premičre intention n'ayant pas fait leur preuve [10,11]. Cette tumeur ne répond pas ŕ la chimiothérapie et les résultats concernant la radiothérapie sont discordants.
Le CMES est une entité rare distincte sur le plan clinique, histologique, immuno-histo-chimique, cytogénétique et évolutif. La clé diagnostique est morphologique, aidée par l'immuno-histochimie et l'étude génétique t (9;22). C'est une tumeur de diagnostic trčs difficile et souvent retardé, et malgré son agressivité surtout locale et la survie prolongée, elle est considérée comme un sarcome de bas grade de malignité ou de malignité intermédiaire.
Les auteurs ne déclarent aucun conflit d'intéręts dans le cadre de cette étude.
Tous les auteurs ont contribué ŕ cette étude depuis la conception, la lecture, et ont approuvé la derničre version.
Figure 1: IRM en coupe coronale en séquence pondérée T2 montrant une masse tumorale assez bien limitée en hypersignal
Figure 2: architecture lobulée, fond myxoďde, cellules atypiques dans des logettes
Figure 3: vue per-opératoire montrant la résection carcinologique de la tumeur
- Oliveira AM, Sebo TJ, Mc Grogy JE, Gaffey TA, Rock MG, Nascimento AG. Extraskeletal myxoid chondrosarcoma : a clinicopathologic, immunohistochemical and ploidy analysis of 23 cases. Mod Pathol . 2000 ; (13) :900-8. PubMed | Google Scholar
- Okamoto S, Hisaoka M, Ishida T, et al . Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and molecular analysis of 18 cases. Hum Pathol . 2001 ; 32(10) :1116-24. PubMed | Google Scholar
- McGrory JE, Rock MG, Nascimento AG, et al. Extraskeletal myxoid chondrosarcoma. Clin Orthop . 2001 ; (382):185-90. PubMed | Google Scholar
- Kempson RL, Fletcher CDM, Evans HL, et al. Extraskeletal myxoid chondrosarcoma. Tumours of soft tissue. 2001; AFIP: 400-5. PubMed | Google Scholar
- Mills SE, Gaffey MJ, Frierson HF. Tumours of the upper aerodigestive tract and ear. AFIP Atlas of tumour pathology. 2000; Third series, fascicle 30. Google Scholar
- Lucas DR, Heim S. Extra skeletal myxoid chondrosarcoma, In: Fletcher CDM, Unni KK, Mertens F. Tumours of soft tissue and bone, OMS. 2002; 213-4. PubMed | Google Scholar
- Mizuho Y, Junichi I, Mamoru T, et al .Chondrosarcoma of the temporal bone. Auris Nasus Larynx. 2007; 34(4):527-31. PubMed | Google Scholar
- Meis-Kindblom JM, Bergh P, Gunterberg B, et al. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and pronostic factors based on 117 cases. Am J Surg Pathol. 1999; 23(6): 636-50. PubMed | Google Scholar
- Santacruz MR, Proctor L, Thomas DB, Gehrig PA. Extraskeletal myxoid chondrosarcoma: a report of a gynecologic case. Gynecol Oncol. 2005 ; 98(3) :498-501. PubMed | Google Scholar
- Algros MP, Collonge-Rame MA, Bedgejian I, et al .Différenciation neuroectodermique des chondrosarcomes extrasquelettiques myxoďdes? un classique? Ann Pathol .2003; 23(3) :244-8. PubMed | Google Scholar
- Acero J, Escrig M, Gimeno M, Montenegro T, Navarro-Vila C. Extraskeletal myxoid chondrosarcoma of the infratemporal fossa: a case report. Int J Oral Maxillofac Surg. 2003; 32(3) :342-5. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
