
Le syndrome de Stewart-Treves compliquant un lymphśdčme chronique idiopathique
Fatima Safini , Asmaa Naim, Zineb Bouchbika, Nadia Benchakroun, Hassan Jouhadi, Nezha Tawfiq, Souha Sahraoui, Abdellatif Benider
Corresponding author: Fatima Safini, Service de Radiothérapie-Oncologie, Centre hospitalier Ibn Rochd, Casablanca 1, quartier des hôpitaux 20360, Casablanca, Maroc 
Received: 18 Oct 2014 - Accepted: 01 Nov 2014 - Published: 21 Nov 2014
Domain: Clinical medicine
Keywords: syndrome de Stewart-Treves,lymphoedème,lymphangiosarcome
©Fatima Safini et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Fatima Safini et al. Le syndrome de Stewart-Treves compliquant un lymphśdčme chronique idiopathique. Pan African Medical Journal. 2014;19:311. [doi: 10.11604/pamj.2014.19.311.5636]
Available online at: https://www.panafrican-med-journal.com//content/article/19/311/full
Original article 

Le syndrome de Stewart-Treves compliquant un lymphśdčme chronique idiopathique
Le syndrome de Stewart-Treves compliquant un lymphśdčme chronique idiopathique
Fatima Safini1,&, Asmaa Naim1, Zineb Bouchbika1, Nadia Benchakroun1, Hassan Jouhadi1, Nezha Tawfiq1, Souha Sahraoui1, Abdellatif Benider1
1Service de Radiothérapie-Oncologie, Centre hospitalier Ibn Rochd, Casablanca 1, quartier des hôpitaux 20360, Casablanca, Maroc
&Auteur correspondant
Fatima Safini, Service de Radiothérapie-Oncologie, Centre hospitalier Ibn Rochd,
Casablanca 1, quartier des hôpitaux 20360, Casablanca, Maroc
Le syndrome de Stewart-Treves (SST) est une entité rare, correspondant ŕ un angiosarcome cutané compliquant un lymphoedčme chronique. Il est de mauvais pronostic. Stewart et Treves ont rapportés en 1948, les premiers cas d'angiosarcome secondaire ŕ un traitement du cancer du sein. Ce terme s'est généralisé pour regrouper l'ensemble des cas de lymphangiosarcome sur lymphoedčme d'origine congénital héréditaire ou non héréditaire, post-traumatique ou post-infectieux. Le SST sur un lymphoedčme idiopathique reste exceptionnel. Nous rapportons le cas rare d'une patiente présentant un lymphoedčme chronique primitif idiopathique des quatre membres évoluant depuis l'adolescence et qui a développé un SST du membre supérieur droit. Elle a subi une amputation ŕ mi- bras vu le caractčre trčs évolué de la tumeur.
L'angiosarcome cutané (AS) est une tumeur endothéliale maligne peu fréquente et grave. Il complique le plus souvent un lymphœdčme chronique congénital ou acquis. Stewart et Treves avaient publié en 1948, une premičre série de 6 cas de lymphangiosarcome sur lymphoedčme chronique aprčs mastectomie, curage axillaire et radiothérapie pour un cancer du sein [1]. Depuis cette description, plus de 400 cas de syndrome de Stewart-Treves (SST) ont été rapportés dans la littérature dans un contexte de stase lymphatique chronique d'origine diverse : congénitale [2], post-radique [3], secondaire ŕ une filariose [4] ou ŕ un traumatisme [5]. Nous rapportons une nouvelle observation, une revue de littérature est aussi présentée.
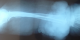
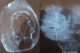
Il s'agit d'une femme âgée de 70 ans, ayant un retard mental connu, qui présente un lymph'dčme chronique des quatre membres évoluant depuis l'adolescence. Pas de cas similaires dans la famille. La patiente a consulté en Décembre 2012, aprčs 3 ans d'évolution d'une lésion nodulaire de la face antérieure du poignet droit augmentant progressivement de taille, évoluant vers la nécrose et l'ulcération. L'examen clinique ŕ l'admission a objectivé une lésion cutanée bourgeonnante et multi-nodulaire d'aspect bleu-grisâtre et hémorragique en périphérie. Ce processus mesurant 17×11×4cm, est situé sur un membre oedématié. La radiographie standard du membre supérieur droit a objectivé un œdčme important des parties molles avec épaississement plus intense au niveau du tiers distal (Figure 1 ). L'IRM du męme membre a mis en évidence un processus papulo-nodulaire de la face antérieure du poignet et de l'avant bras droit, hétérogčne, mal limité, hypointense en T1 et hyperintense en T2 et en séquence de saturation de graisse, avec réhaussement massif et hétérogčne ŕ l'injection du gadolinium. Ce processus bourgeonnant infiltre les parties molles superficielles, sans atteinte des structures osseuses (Figure 2 ). Une biopsie de la lésion a confirmé le diagnostic d'un angiosarcome cutané sur lymphoedčme chronique. L'immunohistochimie a éliminé un sarcome de Kaposi devant la négativité de l'anti- HHV8. Le bilan d'extension (TDM thoracique) était normal. Aprčs confirmation histologique, une chirurgie radicale ŕ type d´amputation ŕ mi-bras droit a été pratiquée vu que la lésion était localement avancée et non métastatique. L'exérčse était complčte avec des marges saines et suffisantes située ŕ 13 cm en distal et 29 cm en proximal avec la limite osseuse ŕ 22cm. Aprčs discussion du dossier en réunion de concertation pluridisciplinaire, une surveillance a été préconisée avec des mesures préventives pour les autres membres. La patiente est toujours sous surveillance aprčs 24mois de recul sans récidive locale ou ŕ distance ni autres nouvelles lésions.
Les angiosarcomes (AS) sont des tumeurs rares, d'évolution péjorative et de mauvais pronostic, représentant environ 1 ŕ 2 % des sarcomes des tissus mous et moins de 1% des tumeurs malignes [6]. Ils surviennent souvent dans le cadre du syndrome de Stewart-Treves (SST), anciennement appelé lymphangiosarcome. C'est une complication rare du lymphoedčme chronique survenant dans 90 % des cas dans un contexte d'antécédent de néoplasie mammaire [1]. Il a été rarement rapporté dans d'autres situations de stase lymphatique telles le lymphoedčme congénital héréditaire ou non héréditaire, post-traumatique ou post-infectieux (filariose) et exceptionnellement idiopathique [2-5,7]. En effet, la majorité des études publiées ont traité le lymphangiosarcome sur lymph'dčme chronique post-thérapeutique d'un cancer du sein alors que les autres causes du lymphoedčme chronique n'avaient fait l'objet que de quelques observations isolées notamment l'angiosarcome compliquant un lymphoedčme primitif idiopathique.
Dans une revue de la littérature réalisée par Woodward, ayant collecté 186 cas de lymphangiosarcomes publiés entre 1948 et 1972, seulement 24 étaient survenus sur un lymphœdčme chronique non tumoral [8]. Le mécanisme de survenue du SST reste mal élucidé. La stase lymphatique chronique est le facteur prédisposant le plus reconnu [7,9]. Le SST a été rapporté surtout chez les femmes, avec une moyenne d'âge de survenue de 62 ans. L'angiosarcome sur lymphoedčme primitif idiopathique a été rapporté surtout au niveau des membres inférieurs, alors que sa survenue sur le membre supérieur sans antécédent de mastectomie avec curage axillaire est extręmement rare [10]. Notre observation, se distingue par le développement d'un angiosarcome du membre supérieur sur un lymphoedčme non rattaché ŕ une néoplasie mammaire. Sur le plan histologique, le diagnostic d'un angiosarcome est souvent difficile vu qu'il présente une double composante vasculaire et lymphatique. L'immuno-histochimie garde une place importante surtout dans les formes peu et indifférenciées. Elle permet de confirmer la nature vasculaire de la prolifération en exprimant les marqueurs UEA-1, CD3, le facteur VIII et le CD34 et d'éliminer les autres diagnostics différentiels. D'autres marqueurs peuvent aider également au diagnostic tels les anti-laminines, le collagčne IV, la vimentine ainsi que l'anti CD31. Le grade histologique qui est un facteur pronostic des sarcomes en général, est difficile ŕ appliquer aux angiosarcomes [10,11].
Malheureusement, peu d'études ont mis le point sur l'apport de l'imagerie dans la prise en charge du SST. Quelques observations ont décrit l'aspect de la tumeur ŕ l'imagerie par résonance magnétique. L'IRM trouve son intéręt surtout dans l'évaluation de l'extension en surface et en profondeur de l'angiosarcome. Elle est plus précise dans l'estimation des marges et du degré d'envahissement des fascias et des muscles sous-jacents. Elle permet d'orienter la biopsie vers la lésion la plus suspecte et de déterminer d'autres lésions satellites [12].Vu la rareté du SST, on ne dispose pas d'une stratégie thérapeutique bien codifiée. Le traitement de référence est la chirurgie radicale. Une excision large est acceptée si elle permet d'obtenir des marges de sécurité saines et suffisantes. Dans les cas oů les lésions sont étendues, comme celui de notre cas, il faut aller jusqu'ŕ l'amputation voire męme une désarticulation pour assurer des marges de sécurité les plus larges possibles. Le risque de récidive locale paraît plus élevé en cas d'exérčse limitée [8, 9,13]. La radiothérapie a montré une certaine efficacité dans le traitement du SST. Elle peut ętre réalisée en premičre intention avant la chirurgie ou en adjuvant [14,15].
La chimiothérapie est indiquée dans les formes localement avancées non opérables et dans les formes métastatiques, męme si son bénéfice reste controversé. Plusieurs agents de chimiothérapie ont été utilisé soit en monothérapie ou en association (5-fluorouracil, methotrexate, bléomycine, vincristine, actinomycin D, doxorubicin, cyclophosphamide et dacarbazine) [16]. Deux équipes japonaises ont publié des résultats controversés sur l'utilisation de l´immunothérapie active par interleukine 2 (IL2) recombinante. Paradoxalement, la place de l´interféron alpha n´a pas encore été évaluée dans le traitement du syndrome de Stewart-Treves, malgré son efficacité dans le traitement de l´hémangiome chez l´enfant et dans le sarcome de Kaposi [17,18].
Vu la nature agressive du SST et le risque élevé de récidives locales et ŕ distance, la survie ŕ long terme est médiocre avec une survie médiane de 2,5 ans aprčs le diagnostic [9]. Il semble que la survie soit meilleure en cas d'origine non tumorale du lymphoedčme chronique comme c'est le cas de notre patiente. Woodward a rapporté une survie médiane de 19 mois pour les cas d'angiosarcome post-mastectomie contre 34 mois pour les cas d'angiosarcome sur lymphoedčme chronique non tumoral [8]. La prévention et le traitement du lymphoedčme chronique demeurent nécessaires et obligatoires dans la lutte contre ce syndrome qui reste de mauvais pronostic malgré sa rareté [9]
Le SST est une complication tardive, rare mais grave des lymphœdčmes chroniques. La précocité de la prise en charge diagnostique et thérapeutique est le seul garant d'une amélioration de la survie. Il est aussi important de souligner l'intéręt d'une surveillance clinique réguličre des patients souffrant d'un lymphoedčme chronique et d'insister sur le respect des mesures préventives. Comme c'est le cas de notre patiente qui présente toujours un lymphoedčme des trois membres nécessitant une surveillance étroite vu le risque de survenue de nouvelles complications malignes notamment le SST au niveau des autres membres.
Les auteurs déclarent n´avoir aucun conflit d´intéręt.
Tous les auteurs ont contribué ŕ la conduite de ce travail. Tous les auteurs déclarent également avoir lu et approuvé la version finale du manuscrit.
Figure 1 : radiographie standard objectivant un śdčme important des parties molles du membre supérieur avec épaississement plus intense au niveau du tiers distal du membre supérieur droit
Figure 2 : IRM des parties molles du membre supérieur objectivant un processus papulo-nodulaire de la face antérieure du poignet et de l’avant bras droit, hétérogčne, infiltrant les parties molles superficielles, sans atteinte des structures osseuses
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948 May;1(1): 64-81. PubMed | Google Scholar
- Wendt I, Kietzmann H, Schubert C, Kaiserling E. Progressive lymphangiokeratoma and angiosarcoma (Stewart et Treves syndrome in congenital lymphedema). Hautartzt .1988 Mar; 39(3): 155-60. PubMed | Google Scholar
- Taghian A, de Vathaire F, Terrier P, Le M, Auquier A, Mouriesse H et al. Long-term risk of sarcoma following radiation treatment for breast cancer. Radiat Oncol Biol Phys. 1991 Jul; 21(2) :361-367. PubMed | Google Scholar
- Shubhangi V Agale, Wasif Ali ZA Khan, and Karishma Chawlani. Chronic Lymphedema of Filarial Origin: A Very Rare Etiology of Cutaneous Lymphangiosarcoma. Indian J Dermatol. 2013 Jan-Feb; 58(1) : 71-73. PubMed | Google Scholar
- Brigitte Laguerre, Claudia Lefeuvre, Pierre Kerbrat, Mohamed Hassel . Syndrome de Stewart-Treves sur lymphœdčme chronique post-traumatique. Bulletin du Cancer. 1999; 86(3): 279-82. PubMed | Google Scholar
- Morel M, Taieb S, Penel N, et al. Imaging of the most frequent superficial soft-tissue sarcomas. Skeletal Radiol. 2011; 40(3):271-84. PubMed | Google Scholar
- Danese C A, Grishman E et al. Malignant Vascular Tumors of the Lymphedematous Extremity. Ann Surg. 1967; 166(2): 245-253. PubMed | Google Scholar
- Woodward AH, Ivins JC, Soule EH. Lymphangiosarcoma arising in chronic lymphedematous extremities. Cancer. 1972; 30(2): 562-72. PubMed | Google Scholar
- Amit Sharma, and Robert A, Schwartz. Stewart-Treves syndrome: Pathogenesis and management. J Am Acad Dermatol. 2012; 67(6) : 1342-1348. PubMed | Google Scholar
- Devolder Sylvette, Breuillard François, Gross Sophie. Lymphangiosarcoma of Stewart-Treves. European Journal of Dermatology. 1998; 8(7):527-8. PubMed | Google Scholar
- Ord onez N, Batsakis J. Comparison of Ulex europaeusI lectin and factor VIII-related antigen in vascular lesions. Arch Pathol Lab Med. 1984; 108(2):129-32. PubMed | Google Scholar
- Chopra S, Ors F, Bergin D. MRI of angiosarcoma associated with chronic lymphoedema: Stewart Treves syndrome. Br J Radiol. 2007; 80 (960):310-3. PubMed | Google Scholar
- Sánchez-Medina MT, Acosta A, Vilar, Fernández-Palacios J. Angiosarcoma en linfedema crónico (síndrome de Stewart-Treves). Actas Dermosifiliogr. 2012; 103(6):545-7. PubMed | Google Scholar
- Di Simone RN, El-Mahdi AM, Hazra T, Lott S. The response of Stewart-Treves syndrome to radiotherapy. Radiology. 1970 Oct; 97(1):121-5. PubMed | Google Scholar
- Lee SB, Cho BK, Houh W, Song YT, Shim SI, Choi IB. A case of Stewart-Treves syndrome. J Korean Med Sci. 1988 Jun;3(2):83-8. PubMed | Google Scholar
- Malhaire J, Labat J, Simon H, Le Maux H, Spindler P, Lucas B, Lamezec B. One case of Stewart-Treves syndrome successfully treated at two years by chemotherapy and radiation therapy in a 73-year-old woman. Acta Oncol. 1997;36(4):442-3. PubMed | Google Scholar
- Nabeya R, Kobayashi M, Fujita M, Yorituji K, Eguchi N, Okamoto S. A case of Stewart-Treves syndrome: Treatment with recombinant interleukin-2 and a review of japanese literature. Nippon Hifuka Gakkai Zasshi. 1990; 100 (10):1029-39. PubMed | Google Scholar
- Furue M, Yamada N, Takahashi I, Kikuchi K, Tsuchida T, Ishibashi Y, et al. Immunotherapy for Stewart-Treves syndrome: Usefulness of intrapleural administration of tumor-infiltrating lymphocytes against massive pleural effusion caused by metastatic angiosarcoma. J Am Acad Dermatol. 1994 May;30(5 Pt 2):899-903. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures
Article metrics
Recently from the PAMJ








Authors´ services
