
Syndrome d’Allgrove découvert sur une anémie ferriprive chez un enfant de 3 ans
Rim Amrani, Anass Es-seddiki, Anass Ayyad, Sahar Messaoudi, Nassira Tazi
Corresponding author: Rim Amrani, Service de Pédiatrie, Centre Hospitalier Universitaire Mohamed VI, Faculté de médecine et de pharmacie d’Oujda, Université Mohammed Premier, Oujda, Maroc 
Received: 29 Sep 2014 - Accepted: 14 Oct 2014 - Published: 28 Oct 2014
Domain: Maternal and child health
Keywords: anémie, Insuffisance surrénale, alacrymie, Syndrome d´Allgrove, mélanodermie, hypoglycémie
©Rim Amrani et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Rim Amrani et al. Syndrome d’Allgrove découvert sur une anémie ferriprive chez un enfant de 3 ans. Pan African Medical Journal. 2014;19:218. [doi: 10.11604/pamj.2014.19.218.5511]
Available online at: https://www.panafrican-med-journal.com//content/article/19/218/full
Original article 

Syndrome d’Allgrove découvert sur une anémie ferriprive chez un enfant de 3 ans
Syndrome d’Allgrove découvert sur une anémie ferriprive chez un enfant de 3 ans
Rim Amrani1,&, Anass Es-seddiki1, Anass Ayyad1, Sahar Messaoudi1, Nassira Tazi2
1Service de Pédiatrie, Centre Hospitalier Universitaire Mohamed VI, Faculté de médecine et de pharmacie d’Oujda, Université Mohammed Premier, Oujda, Maroc, 2Service de Pédiatrie, Hôpital l Farabi, Oujda, Maroc
&Auteur correspondant
Rim Amrani, Service de Pédiatrie, Centre Hospitalier Universitaire Mohamed VI, Faculté de médecine et de pharmacie d’Oujda, Université Mohammed Premier, Oujda, Maroc
Nous rapportons l'observation d'un enfant de 03 ans, qui s'est présenté en consultation pour une anémie hypochrome microcytaire ferriprive traînante 18 mois auparavant, et qui a été mis sous traitement symptomatique (fer) mal suivi en ambulatoire. Au cours de l'examen clinique, nous avons découvert une pigmentation péri buccale, de la face dorsale des mains, des plis de flexion palmaires, des organes génitaux externes, des pieds, ainsi que des macules pigmentées au niveau des deux jambes, apparue 6 mois auparavant et passée inaperçue. Devant ce tableau clinique, un bilan biologique a montré des hypoglycémies, un taux de Cortisol de 8 heures du matin effondré, un taux de Rénine bas, un taux d'ACTH élevé, un test au Synacthčne® négatif, ce qui nous a permis d'évoquer une insuffisance surrénalienne. Un bilan étiologique a été réalisé dont un bilan immunologique avec un dosage de la 17 hydroxy-progestérone, des anticorps anti 21 hydroxylase, des anticorps anti-thyroglobuline et des acides gras ŕ trčs longues chaines, et qui est revenu normal. Le bilan infectieux et radiologique a été normal. Une alacrymie a été suspectée et alors confirmée par un test de Schirmer positif, ce qui rentre dans le cadre d'une forme incomplčte du syndrome d'Allgrove ou syndrome des 3A fait habituellement d'anémie, d'insuffisance surrénalienne et d'achalasie (inconstante).
L'insuffisance surrénale (IS) est une maladie relativement rare (90 ŕ 140 cas par million d'habitants) [1], mais grave si elle n'est pas diagnostiquée et traitée vu le risque d'évolution vers l'insuffisance surrénale aiguë qui peut ętre mortelle. L'insuffisance surrénale peut ętre primaire et alors le plus souvent due ŕ une lésion auto-immune, génétique ou ŕ une tuberculose. Elle peut ętre secondaire ŕ un déficit corticotrope et alors secondaire ŕ une corticothérapie ou ŕ une lésion hypothalamo-hypophysaire [1,2]. Nous rapportons le cas d'un enfant de 3 ans, ayant présenté une insuffisance surrénale révélée par une mélanodermie isolée et une anémie associée ŕ une alacrymie, faisant découvrir un syndrome d'Allgrove.
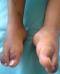
Il s'agit d'un enfant âgé de 3 ans, avec des antécédents d'anémie ferriprive évoluant depuis 18 mois, suivi en ambulatoire, ayant consulté pour persistance de l'anémie chez qui on a découvert une pigmentation cutanée apparue 6 mois auparavant et passée inaperçue. Le patient n'a pas présenté d'asthénie mais une stagnation pondérale ŕ 10 kg pendant plus de 12 mois avec moins 4 déviations standards (DS) sur le poids, moins 3 DS sur la taille et une tension artérielle correcte. L'examen a montré une pigmentation péri buccale, de la face dorsale des mains, des plis de flexion palmaires, des organes génitaux externes et des pieds, ainsi que des macules pigmentées au niveau des deux jambes (Figure 1, Figure 2, Figure 3). La numération formule sanguine a montré un taux d'hémoglobine ŕ 8.9g/dl, un volume globulaire moyen (VGM) ŕ 68fl, une concentration corpusculaire moyenne en hémoglobine (CCMH) ŕ 28.6 g/dl et une ferritinémie ŕ 6.61 nanogramme (ng)/ml, les chiffres glycémiques ont varié entre 0.45g/l ŕ l'admission et 1,20 g/l, une natrémie ŕ 142 mmol/l et une kaliémie ŕ 4,8 mmol/l. La cortisolémie de base a été trčs effondrée ŕ 3,55 microgramme (µg)/dl, la Rénine ŕ 2.50 picogramme (pg)/l, le taux d'ACTH plasmatique élevé ŕ 354 pg/ml, le cortisol plasmatique dosé 60 minutes aprčs l'injection intramusculaire de 0.25 mg de tétracosactide (Synacthčne®) a été de 3,72 microgramme µg/dl.
Le diagnostic d'une insuffisance surrénale primitive ou maladie d'Addison a été ainsi retenu. La fonction rénale et hépatique ont été correctes. Le bilan thyroďdien (TSH- T4- T3) a été normal ainsi que le bilan phosphocalcique (calcémie- phosphorémie- parathormone). Le dosage des anticorps anti-transglutaminases Ig G a été normal. Le bilan infectieux a été négatif (tuberculose- CMV et sérologie HIV). Un bilan immunologique dont le dosage de la 17 hydroxy-progestérone (0.10 ng/l), des anticorps anti 21 hydroxylase (0.92 U/ml), des anticorps anti-thyrogloguline (< 70UI/l) et des acides gras ŕ trčs longues chaines a été normal ainsi que le bilan radiologique dont l'âge osseux, la radiographie de poumon, l'échographie abdominale, la TDM abdominale et l'IRM cérébrale. L'enquęte étiologique de l'IS a été complétée par un examen ophtalmologique ŕ la recherche du syndrome d'Allgrove (ou syndrome des 3A) .Dans ce sens, un interrogatoire poussé avec les parents, a retrouvé une alacrymie qui a été négligée, et qu'on a confirmé par un test de Schirmer, mais sans achalasie. L'enfant a été traité par hydrocortisone ŕ la dose de 10 mg/m˛/j, avec une nette amélioration clinique (Figure 4), une correction de sa glycémie et une reprise pondérale. Il a été mis en parallčle sous traitement par des larmes artificielles. Par ailleurs, l'anémie s'est corrigée rapidement au bout d'un mois du début traitement. L'enfant est suivi en consultation, avec un contrôle régulier de ses chiffres tensionnels et de sa glycémie. Une carte d'insuffisant surrénalien lui a été remise.
L'insuffisance surrénale lente primitive chronique ou maladie d'Addison est liée ŕ un dysfonctionnement de la glande surrénale. Elle comporte un déficit en hormones surrénaliennes [3,4]. Chez cet enfant, malgré l'absence de signes généraux (asthénie et hypotension artérielle) et l'existence d'une mélanodermie qui est caractéristique de la maladie d'Addison, l'insuffisance surrénale a été recherchée devant la localisation de la pigmentation aux régions découvertes et aux plis de flexion, ainsi que la présence de tâches ardoisées et l'hypoglycémie découverte ŕ l'admission [3, 5, 6]. La rareté de la pathologie et le caractčre non spécifique de l'asthénie peuvent retarder le diagnostic ; ce qui a été le cas de notre patient qui a été suivi pour son anémie pendant 18 mois sans que le diagnostic ne soit posé. La mélanodermie a des caractéristiques spécifiques ; elle est différente de la pigmentation de l'hémochromatose et de la porphyrie cutanée tardive [3]. Une pigmentation cutanéo-muqueuse peut aussi ętre observée lors de l'exposition chronique ŕ des métaux (mercure, plomb, argent, etc.) [3]. Les dosages hormonaux (cortisol, ACTH, rénine et le test au Synacthčne®) ont confirmé notre diagnostic [6-8]. Les désordres électrolytiques (hyponatrémie et hyperkaliémie habituellement rencontrés dans l'IS chez l'enfant [1,4]), n'ont pas été notés chez notre patient. Le diagnostic de l'insuffisance surrénale aurait pu alors rester méconnu et n'aurait été fait que devant un tableau d'insuffisance surrénale aigüe.
Chez cette enfant, le bilan tuberculeux a été négatif; étiologie classiquement retrouvée dans un pays endémique comme le Maroc, la sérologie CMV et VIH ont été négatives. Le dosage des anticorps anti-transglutaminases, devant le retard staturo-pondéral et l'anémie trainante, a été normal. L'exploration de la fonction thyroďdienne et parathyroďdienne ŕ la recherche d'endocrinopathie a été normale. Le taux de la 17 hydroxy-progestérone a été normal éliminant ainsi l'hyperplasie congénitale des surrénales qui représente l'une des étiologies de l'IS chez le nourrisson et l'enfant [4]. Le dosage des anticorps anti-thyroglobuline a été normal ainsi que le taux d'anticorps anti 21 hydroxylase éliminant ainsi les causes auto-immunes de l'IS [3, 4, 6]. Le dosage des acides gras ŕ trčs longues chaines dans le cadre de l'adrénoleucodystrophie (ADL); retrouvée parmi les étiologies de l'IS, a été normal [6, 9]. L'enquęte étiologique de l'IS a été complétée par un examen ophtalmologique ŕ la recherche du syndrome d'Allgrove (ou syndrome des 3A) retrouvé dans 50% des cas d'IS chez l'enfant [1] .Dans ce sens, un test de Schirmer a été positif confirmant l'alacrymie, mais sans achalasie qui n'est habituellement retrouvée que dans 75% des cas de syndrome d'Allgrove [1, 2, 6]. L'étude génétique n'a pas été réalisée. Le bilan radiologique a été sans particularité.
Le syndrome d'Allgrove ou syndrome des 3A est une maladie autosomique récessive rare (lié ŕ des mutations du gčne AAAs codant la protéine appelée ALADIN), de prédominance masculine, caractérisée par une alacrymie ; la manifestation la plus précoce de la maladie, une insuffisance surrénalienne (adrenal insufficiency) et une achalasie de l''sophage qui reste le trouble le plus fréquemment rencontré aprčs l'alacrymie [10]. Des troubles neurologiques (neurologic abnomalities) peuvent apparaître ŕ un moment variable de l'évolution de la maladie définissant alors pour certains auteurs le syndrome des 4A [1 ,2 ,10].
Le traitement hormonal a pour but de substituer les déficits en cortisol. Pour cet enfant, nous avons constaté la normalisation de l'hémogramme 1 mois aprčs le début du traitement par Hydrocortisone ; une probable hypothčse physiopathologique sur le rôle du cortisol dans l'hématopoďčse reste ŕ vérifier. L'Hydrocortisone comprimé ŕ la dose de 10 mg/m˛/j répartie en deux prises [3-5] est le seul traitement disponible en attendant les nouvelles molécules ŕ libération prolongée DuoCort® et ChronoCort® dont le but est l'amélioration de la qualité de vie et la diminution de la morbidité chez les insuffisants surrénaliens [7].
Męme isolée, la mélanodermie doit faire évoquer l'insuffisance surrénale et faire les explorations hormonales nécessaires, afin d'éviter de graves complications. Les insuffisances surrénales primaires auto-immunes sont désormais les plus fréquentes au sein d'un éventail d'étiologies qui se diversifie progressivement (formes génétiques dont le syndrome d'Allgrove ou ADL, sida, tuberculose..). Par ailleurs, les bases du traitement reposent sur la substitution en glucocorticoďdes et si nécessaire en minéralocorticoďdes, ainsi que sur l'éducation du patient qui participe activement ŕ la prévention des accidents aigus.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la réalisation de ce travail. Tous ont lu et approuvé la version finale du manuscrit.
Figure 1: pigmentation
péri buccale
Figure 2:
pigmentation de la face dorsale des mains et des plis de flexion
palmaires
Figure 3:
macules pigmentées au niveau des deux jambes
Figure 4: l'enfant
3 mois aprčs le début du traitement
- Matoussi N, Amdouni N, Fitouri Z, Makni S, Cheour M et al. Clinical and etiological features of primary adrenal insufficiencies in children. Tunis Med. 2008; 86(10):890-894. PubMed | Google Scholar
- Louati H, Rebah A, Douira W, Debabria H, Sayed M, Ghorbel S, et al. Apport de l’imagerie dans le syndrome des 3A .Journal de Radiologie. 2007 ; 88 (10): 1595. PubMed | Google Scholar
- Oliver C. Insuffisance surrénale. EMC. 2009; (3): 0540. PubMed | Google Scholar
- Raux Demay M-C. Corticosurrénales: physiopathologie et explorations Endocrinologie. EMC. 2010; (4): 107-A-15. Google Scholar
- Raux Demay M-C. Endocrinologie, Pathologie des corticosurrénales. EMC. 2010 ; (4): 107-A-20. Google Scholar
- Menon S, Kuhn J-M. Insuffisance surrénalienne. EMC Endocrinologie-Nutrition. 2011 ; (10): 015-A-10. Google Scholar
- Nunes M-L, Tabarin A. Les actualités de l’insuffisance surrénalienne (News for adrenal insufficiency). Annales d’Endocrinologie. 2008 ; 69(1): 44-52. PubMed | Google Scholar
- Badri T, Zeglaoui F, Khiari K, et al .Pigmentation cutanée isolée: penser ŕ l’insuffisance surrénale. Presse Med. 2007 ; 36 (4 Pt 1): 615-8. PubMed | Google Scholar
- Hsieh S, White P-C. Presentation of Primary Adrenal Insufficiency in Childhood. J Clin Endocrinol Metab. 2011; 96 (6): 925-928. PubMed | Google Scholar
- Ben Fredj Ismail F, Ben Slamab A, Mzabia A et al. Syndrome d’Allgrove ŕ révélation tardive. Annales d’Endocrinologie. 2012 ; 73 (4) : 336-353. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures



Article metrics
Recently from the PAMJ








Authors´ services
