
Paragangliome rétropéritonéal non secrétant: une cause rare d’occlusion intestinale haute
Mehdi Soufi, Said Benamr, Bouziane Chad
Corresponding author: Mehdi Soufi, Département de Chirurgie, Faculté de Médecine d’Oujda, Université Mohammed Ier Oujda, Maroc 
Received: 25 Jul 2014 - Accepted: 10 Aug 2014 - Published: 20 Aug 2014
Domain: Clinical medicine
Keywords: Paragangliome, tumeur rétropéritonéal, chirurgie, concertation multidisciplinaire.
©Mehdi Soufi et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Mehdi Soufi et al. Paragangliome rétropéritonéal non secrétant: une cause rare d’occlusion intestinale haute. Pan African Medical Journal. 2014;18:312. [doi: 10.11604/pamj.2014.18.312.5140]
Available online at: https://www.panafrican-med-journal.com//content/article/18/312/full
Original article 

Paragangliome rétropéritonéal non secrétant: une cause rare d’occlusion intestinale haute
Paragangliome rétropéritonéal non secrétant: une cause rare d’occlusion intestinale haute
Mehdi Soufi1,&, Said Benamr2, Bouziane Chad2
1Département de Chirurgie, Faculté de Médecine d’Oujda, Université Mohammed Ier Oujda, Maroc, 2Clinique chirurgicale B Avicenne Rabat, Rabat, Maroc
&Auteur correspondant
Mehdi Soufi, Département de Chirurgie, Faculté de Médecine d’Oujda, Université Mohammed Ier Oujda, Maroc
Les paragangliomes sont des tumeurs neuroendocrines rares diagnostiquées le plus souvent chez le jeune adulte. Nous rapportons le cas rare d'une jeune femme de 20 ans présentant un paragangliome rétropéritonéal situé sur la face antérolatérale de l'aorte et comprimant l'angle duodéno-jéjunal responsable d'un syndrome subocclusif haut. Dans les formes non sécrétantes, la symptomatologie est souvent déroutante. Le diagnostic n'est généralement fait qu'aprčs examen histologique de la pičce de résection. Le traitement de choix repose sur une chirurgie carcinologique intégrant une concertation multidisciplinaire.
Les paragangliomes (PG) ou phéochromocytomes extra-surrénaliens sont des tumeurs rares développées aux dépens des cellules neuro-ectodermiques du systčme nerveux autonome [1]. Moins de 2% se localisent en rétropéritonéal. La forme non sécrétante est exceptionnelle [2]. Elle reste longtemps asymptomatique, ce qui amčne ŕ la découvrir ŕ des stades avancés [2]. A notre connaissance, La compression de l'angle duodéno-jéjunal est un mode de révélation jamais rapporté. Malgré les progrčs de l'imagerie, cette forme pose un problčme diagnostique qui n'est le plus souvent confirmé qu'aprčs l'analyse histologique de la pičce opératoire [3]. Nous rapportons un nouveau cas dont la symptomatologie était révélée par un syndrome compressif haut chez une jeune fille de 20 ans. A travers cette observation, nous discuterons certains problčmes diagnostiques et thérapeutiques que posent ces tumeurs non sécrétantes.
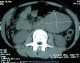
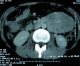
Mlle E.F âgée de 20 ans, sans antécédents particuliers, avait consulté pour des douleurs para ombilicales gauches associées ŕ des vomissements incoercibles post prandiaux tardifs d'évolution croissante. Il n'y avait pas de fičvre ni d'hémorragie digestive. Cette symptomatologie évoluait dans un contexte d'amaigrissement chiffré ŕ 13 kilos en 1 mois. L'examen retrouvait une patiente amaigrie avec plis de déshydratation et de dénutrition. L'abdomen, souple, était le sičge d'une masse latéro ombilicale sensible, fixe par rapport au plan profond d'environ de 10 cm de diamčtre. Le reste de l'examen était sans particularité. Une échographie révélait l'existence en latéro aortique gauche d'une formation hétérogčne de 8,5 x 6 cm de diamčtre. Le tomodensitométrie (TDM) abdominale, réalisée avant et aprčs injection du produit de contraste, objectivait la présence d'une tumeur occupant l'hypochondre et le flanc gauche, bien limité mesurant 9 x 7 cm, prenant le contraste et ne contenant pas de calcifications ( Figure 1, Figure 2). La radiographie du thorax était sans particularité.
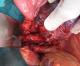
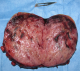
La fibroscopie œsogastroduodénal progressant jusqu' au deuxičme duodénum était normale. En biologie, il existait des signes de déshydratation : une insuffisance rénale fonctionnelle, une hyponatrémie, une hypokaliémie ainsi qu'une hypoprotidémie. Devant ce tableau d'occlusion haute avec retentissement hydroélectrolytique et la présence d'une masse abdominale, une intervention chirurgicale a été indiquée. L'exploration par une laparotomie médiane retrouvait une tumeur rétropéritonéale d'environ 10 cm de diamčtre. Cette tumeur était ferme, rosâtre ŕ surface hypervascularisée reposant sur la face antérolatérale gauche de l'aorte. La masse refoulait et comprimait l'angle duodéno-jéjunal (Figure 3). Une tumorectomie était réalisée aprčs libération du tube digestif et contrôle vasculaire (il existait une branche artérielle efférente de l'aorte abdominale) (Figure 4). Les suites étaient simples. La patiente a quitté l'hôpital ŕ j +2. L'histologie confirmait qu'il s'agissait d'un paragangliome sans signes de malignité (Figure 5). Le suivi clinique et biologique était satisfaisant. Le recul est de deux ans. Une enquęte familiale n'a révélé aucune anomalie génétique particulaire.
Les paragangliomes sont des tumeurs rares qui se développent ŕ partir des cellules germinales de la cręte neurale, et se localisent habituellement au niveau de la médullosurrénale [1]. Moins de 10 % sont extra surrénaliens. Elles peuvent ętre associés ŕ d´autres pathologies rentrant dans le cadre des néoplasies endocriniennes multiples NEM 2, ŕ une maladie de Von hippel-Lindau (VHL) et ŕ une neurofibromatose de type 1 (maladie de Recklinghausen) [4]. En outre, elles peuvent compléter la triade de Carney associant un léiomyosarcome gastrique et un chondrome pulmonaire [5]. Les localisations ectopiques sont le plus souvent en intra abdominales touchant les chaînes sympathiques pelviennes, l'organe de Zuckerkland, la vessie, et męme le rectum et le sacrum. Le paragangliome rétropéritonéal est inhabituel. Il touche le plus souvent l'adulte entre 30 et 50 ans [1,4].
Notre observation présente deux particularités, l'occlusion haute par le syndrome tumoral et l'absence de syndrome endocrinien. Ces tumeurs sont non sécrétantes dans 40 % des cas, ce qui explique ŕ la fois l'absence de signe fonctionnel spécifique (triade de Ménard, sueurs, pas de céphalées, tachycardie, hypotension) [2]. Le plus souvent les formes rétropéritonéales non sécrétantes sont asymptomatiques et peuvent ętre révélées ŕ des stades trčs avancés. Toutefois, des signes d'emprunt, ŕ type de lombalgies, de pesanteur abdominale ainsi que des signes urinaires, ont été décrits [3]. Aucun cas n'a été révélé par un syndrome subocclusif haut suite ŕ une compression de l'angle duodéno-jéjunal. En préopératoire, aucun examen ne nous a permis d'orienter le diagnostic de paragangliome. Dans cette localisation, l'imagerie pose le diagnostic de tumeur rétropéritonéale. L'échographie montre une lésion d'écho structure tissulaire, le plus souvent hétérogčne contenant des zones liquidiennes, ainsi que certaines calcifications [6]. Les paragangliomes ont généralement un aspect scannographique identique ŕ celui des phéochromocytomes surrénaliens, mais leur topographie conditionne la difficulté diagnostique [7]. Chez notre patiente, la masse prenait légčrement le contraste au scanner. L'aspect TDM le plus classique est celui d'une masse bien limitée de plus de 2 cm de diamčtre (souvent entre 4 et 5 cm), massivement rehaussée par le produit de contraste [8,9]. L'imagerie précise aussi l'extension locale de la tumeur, et permet d'évaluer son extirpabilité [7-9].
Dans notre observation, il n'y avait pas de calcifications qui pouvaient orienter le diagnostic. En plus, l´absence de signes d´hypersécrétion de catécholamines, l'indication ŕ réaliser une scintigraphie ŕ la méta-iodo-benzyl-guanidine (MIBG) en préopératoire est inadaptée. Pourtant, cet examen serait positif dans beaucoup de paragangliomes non fonctionnelles et trouve une place dominante dans la surveillance post opératoire ultérieure [2,3]. L'IRM et récemment le Petscan sont trčs sensibles pour détecter les petites localisations infra radiologiques, les métastases de petite taille et les autres lésions ectopiques [10]. Ces examens n'ont pas été réalisés chez notre patiente, il en est de męme pour les examens biologiques. Ces derniers confirment le diagnostic en cas de tumeur sécrétante [1]. Leur dosage était négatif lors de la surveillance postopératoire de notre patiente. Si l'histologie et particuličrement l'immunohistochimie confirment le diagnostic, ils ont un intéręt pronostic grâce au marquage de la protéine S [11]. Le diagnostic de malignité est trčs discuté, puisque seule l´apparition de métastases dans un site oů il n´existe pas habituellement de tissu paraganglionnaire ou la survenue d´une récidive locale ou ŕ distance affirmera la malignité [12].
Le traitement du paragangliome doit rentrer dans un cadre multidisciplinaire. La chirurgie complčte sans résidu microscopique reste le seul traitement qui permet des taux de survies de plus de 75 % ŕ cinq ans [11]. Nous avons réalisé une laparotomie, vu la grande taille de la tumeur. Des auteurs ont rapporté la faisabilité de la coelioscopie dans les tumeurs de moins de 5 cm [13]. La radiothérapie et la chimiothérapie ont permis de contrôler dans quelques cas la maladie chez des patients métastatiques, mais le pronostic dépend de la récidive locale et des marges de résection lors de l'exérčse de la tumeur [14]. 10% des paragangliomes sont héréditaires. En effet, une mutation SDHB (succinate déshydrogénase complex subunit B) s'accompagnait d'une agressivité et d'un risque de récidive plus important que chez les patients ayant une mutation SDHD. Ces données incitent qu'une enquęte génétique soit réalisée systématiquement afin de rechercher ces mutations [15].
Le paragangliome rétropéritonéale est une entité rare. Le mode de révélation par un syndrome subocclusif haut secondaire ŕ la compression de l'angle duodéno-jéjunal est exceptionnel et déroutant surtout en cas de tumeur non sécrétante. L'intéręt de l'imagerie est primordial pour le bilan préthérapeutique. Malgré les progrčs de la radiologie, dans les formes rétropéritonéales non sécrétantes, le diagnostic est souvent affirmé qu'aprčs analyse histologique de la pičce opératoire. Il faut donc savoir l'évoquer devant toute masse rétro péritonéale isolée afin d'entreprendre les précautions nécessaires pour éviter des complications qui peuvent ętre fatales. Le traitement doit intégrer une équipe multidisciplinaire incluant endocrinologue, anatomopathologiste et chirurgien. Seule la chirurgie curative permet de prolonger la survie. Un suivi postopératoire prolongé est recommandé afin de détecter les formes malignes.
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont contribué ŕ la réalisation du manuscrit. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: Scanner sans injection du produit de contraste montrant une tumeur occupant bien limité mesurant 9 x 7 cm prenant le contraste et ne contenant pas de calcifications
Figure 2: Scanner avec injection du produit de contraste montrant la prise de contraste hétérogčne de la tumeur
Figure 3: Image peropératoire aprčs section du muscle de Treitz montrant le paragangliome rétropéritonéal
Figure 4: Pičce de résection ouverte
Figure 5: Aspect histologique du paragangliome
- Barfield R, Hill DA, Hoffer FA, Tekautz T, Spunt SL. Retroperitoneal paraganglioma. Med Pediatr Oncol. 2002; 39(2): 120-4. PubMed | Google Scholar
- Ouaďssi M, Sieleznieff I, Pirrň N, Payan MJ, Chaix JB, Consentino B, Sastre B. Paragangliome non sécrétant rétropéritonéal: Ŕ propos d'une observation. Gastroentérologie Clinique et Biologique. 2007; 31(3): 307-308. PubMed | Google Scholar
- Louafy L, Lakhloufi A, Hamdaoui R, Chehab F, Khaiz D Bouzidi A. Non-functional retroperitoneal paraganglioma. Prog Urol. 2001; 11(3): 512-516. PubMed | Google Scholar
- Dannenberg H, De Krijger RR, Van der Harst E, Abbou M, IJzendoorn Y, Komminoth P. Von Hippel-Lindau gene alterations in sporadic benign and malignant pheochromocytomas. Int J Cancer. 2003; 105(2): 190-195. PubMed | Google Scholar
- Sans N, Durand G, Giron J, Fajadet P, Sénac JP. Triade de Carney, Mise au point: ŕ propos d'un cas. J Radiol. 2000; 81(1) : 39-42. PubMed | Google Scholar
- Giudicelli T, Bruneton JN, Duplay H, Abbes M, Balu-Maestro C, Raffaelli C, Rogopoulos A, Bittmann O. Imagerie des paragangliomes rétropéritonéaux non fonctionnels: A propos d'un cas. J Radiol. 1991; 72(11): 617-619. PubMed | Google Scholar
- Hayes WS, Davidson AJ, Grimley PM, Hartman DS. Extraadrenal retroperitoneal paraganglioma: clinical, pathologic and CT findings. AJR. 1990; 155(6): 1247-1250. PubMed | Google Scholar
- Otal PH, Grenier N, Chabbert V, Basseau F, Joffre F. Imagerie des tumeurs de la surrénale. J Radiol. 2002 (6 Pt 2); 83:897-909. PubMed | Google Scholar
- Chabbert V, Otal PH, Colombier D, Chamontin B, Caron PH, Escourrou G, Rousseau H, Joffre F. Imagerie des phéochromocytomes et des paragangliomes. Feuillet de Radiologie. 2000; 40(2): 107-21. PubMed | Google Scholar
- Puche P, Jacquet E, Colombo PE, Jaber S, Alric P, Carabalona JP, Bouyabrine H, Domergue J, Navarro F. Prise en charge chirurgicale d'un paragangliome préaortique : ŕ propos d'un cas. Annales de Chirurgie. 2006; 131(9): 559-563. PubMed | Google Scholar
- Montresor E, Iacono C, Nifosi F, Zanza A, Modena S, Zamboni G, Bernardello F, Serio G. Retroperitoneal paragangliomas: role of immunohistochemistry in the diagnosis of malignancy and in assessment of prognosis. Eur J Surg. 1994; 160 (10):547-52. PubMed | Google Scholar
- Matsui H, Ikeuchi S, Onoda N, TsutsumiY. Malignant Paraganglioma of the Retroperitoneum with Lung Metastases: A 13-year Survivor After Radical Surgery. Asian journal of surgery. 2007; 30(1): 75-79. PubMed | Google Scholar
- Edstrom Elder E, Hjelm Skog AL, Hoog A, Hamberger B. The management of benign and malignant pheochromocytoma and abdominal paraganglioma. Eur J Surg Oncol. 2003; 29(3):278-83. PubMed | Google Scholar
- Taue R, Takigawa H, Sinotou K, Uno S, Mori R, Tatara K, Sano T. A case of pelvic malignant paraganglioma. Int J Urol. 2001; 8(12): 715-8. PubMed | Google Scholar
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X, COMETE Network. Mutations in the SDHB gene are associated with extraadrenal and/or malignant phaeochromocytomas. Cancer Res. 2003; 63(17): 5615-21. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ








Authors´ services
