
Dystrophie maculaire de Stargardt - ŕ propos d’un cas
Aziz El Ouafi, Med Elmellaoui, Abdelkader Lakataoui
Corresponding author: Aziz El Ouafi, Service d’Ophtalmologie, Hôpital Militaire Moulay Ismail, Mekhnčs, Maroc 
Received: 22 Jun 2014 - Accepted: 05 Jul 2014 - Published: 16 Jul 2014
Domain: Clinical medicine
Keywords: Maculopathie, héréditaire, Stargardt
©Aziz El Ouafi et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Aziz El Ouafi et al. Dystrophie maculaire de Stargardt - ŕ propos d’un cas. Pan African Medical Journal. 2014;18:222. [doi: 10.11604/pamj.2014.18.222.4871]
Available online at: https://www.panafrican-med-journal.com//content/article/18/222/full
Original article 

Dystrophie maculaire de Stargardt - ŕ propos d’un cas
Dystrophie maculaire de Stargardt - ŕ propos d’un cas
Aziz El Ouafi1,&, Med Elmellaoui1, Abdelkader Lakataoui1
1Service d’Ophtalmologie, Hôpital Militaire Moulay Ismail, Mekhnčs, Maroc
&Auteur correspondant
Aziz El Ouafi, Service d’Ophtalmologie, Hôpital Militaire Moulay Ismail, Mekhnčs, Maroc
La maladie de Stardardt est une dystrophie maculaire héréditaire rare d'apparition précoce caractérisée par une perte progressive de la vision centrale, mais avec une vision périphérique intacte, une légčre perte de la vision des couleurs, une adaptation ŕ l'obscurité retardée, et une atrophie maculaire avec ou sans taches paramaculaires et une dégénérescence de l'épithélium pigmentaire de la rétine.
La maladie de Stargardt est la forme la plus fréquente de dystrophie maculaire juvénile ŕ transmission autosomique récessive caractérisée par l'existence d'une lésion maculaire bilatérale ayant un aspect en œil de bœuf en rapport avec l'accumulation de pigment lipofiscine au niveau l'épithélium pigmentaire.
Un garçon âgé de 15 ans se plaint d'une baisse d'acuité visuelle progressive depuis 3 ans. L'acuité visuelle est chiffrée ŕ 1/10 non améliorable en ODG. L'examen du segment antérieur aux deux yeux est sans particularités. L'examen du fond d'œil a révélé l'existence d'une lésion maculaire jaunâtre arrondie avec atrophie maculaire majeure. Le vitré est transparent, l'examen de la périphérie rétinienne est normal.
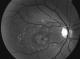
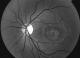
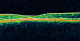
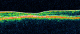
Les clichés en auto fluorescences décrivent un aspect granulaire des maculas avec une alternance de points hypoautofluorescents (altération de l'épithélium pigmentaire) et hyperautofl uorescents (dépôts de lipofuscine). Ces lésions sont accompagnées de taches flavimaculées, spiculées et maculaires (Figure 1, Figure 2). L'angiographie ŕ la fluorescéine confirme la présence d'une atrophie maculaire accompagnée d'un remaniement de l'épithélium pigmentaire. Il existe un effet fenętre comme œuf sur plat au niveau maculaire. L'OCT montre une atrophie maculaire bilatérale majeure, avec disparition de la ligne ellipsoďde (Figure 3, Figure 4). L'électrorétinogramme est normal (Figure 5, Figure 6). L'électroculogramme est normal (Figure 7). L'ensemble de ces examens paracliniques a permis de conclure au diagnostic de maladie de Stargardt de type 1.
La maladie de Stargardt est une maculopathie bilatérale symétrique avec dépôts pisciformes jaunes autofl uorescents [1]. Cette maladie héréditaire est ŕ transmission autosomique récessive. Elle est liée au gčne ABCA4, qui code pour une protéine permettant le transfert du tout-trans-rétinal de la lumičre des disques des photorécepteurs dans leur cytoplasme. L'absence de cette protéine conduit l'accumulation de produits de dégradation du photopigment, toxique pour l'épithélium pigmentaire [2]. Cette maladie affecte surtout les enfants et les adolescents, en provoquant une baisse d'acuité visuelle progressive, accompagnée d'un scotome central et de dyschromatopsies. Le fond d'œil évolue au cours du temps et retrouve de maničre précoce une altération isolée du reflet fovéolaire, oblongue, en oeil de boeuf. Il peut exister, conjointement, des taches pisciformes qui ne touchent pas la zone péripapillaire. Apparaît ensuite, tardivement, une atrophie maculaire Le diagnostic peut se faire uniquement sur les clichés autofluorescents, qui retrouvent une macula hétérogčne (hypo-/hyperautofl uorescente) accompagnée de taches spiculées, hyperautofluorescentes. L'angiographie ŕ la fl uorescéine, non nécessaire au diagnostic, montre un silence choroďdien (de Bonin), présent dans 80 % des cas, ce qui permet d'éliminer certains autres diagnostics (maladie de Stargardt de transmission autosomique dominante, dystrophie des cônes, etc.).[3] L'OCT peut retrouver de maničre précoce une atteinte de la ligne ellipsoďde sur la zone maculaire péri fovéale et un remaniement maculaire atrophique avec diminution de la couche des photorécepteurs. [4] L'électrorétinogramme global est classiquement normal (maladie de Stargardt de type 1) mais peut retrouver, dans certains cas, une altération des réponses scotopiques (type 2) ou photopiques (type 3), entraînant un risque d'atteinte périphérique. Le pronostic est médiocre dés que l'acuité visuelle chute au dessous de 6/10 et elle a tendance ŕ diminuer. Il n'y a pas encore de traitement spécifique pour ces patients ; des essais cliniques de thérapie génique devraient débuter dans les années ŕ venir [3,4].
La dystrophie maculaire de stargardt est une maladie héréditaire ŕ transmission récessive, elle est bilatérale d installation progressive. Le diagnostic se base sur l'interrogatoire le fond d œil et les examens complémentaires (clichés auto fluorescents angiographie OCT et ERG).
Les auteurs ne déclarent aucun conflit d'intéręts.
Tous les auteurs ont participé ŕ l'élaboration de ce travail. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: Atrophie maculaire et remaniements pigmentaires au niveau de l’śil droit
Figure 2: Cliché anerythre OG: aspect granulaire de la macula avec altération de l’épithélium pigmentaire
Figure 3: OCT de l'oeil droit: atrophie maculaire avec disparition de la ligne ellipsoďde
Figure 4: OCT de l'oeil gauche: diminution de l’épaisseur maculaire avec disparition de l’entonnoir foveolaire. Diminution de la couche des photorécepteurs
Figure 5: ERG full field normal
Figure 6: ERG multifocal normal
Figure 7: EOG normal
- Stargardt K. Über familiäre, progressive Degeneration in der Makulagegend des Auges. Graefes Arch Clin Exp Ophthalmol. 1909;71:534-50. PubMed | Google Scholar
- Allikmets R, Singh N, Sun H et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236-46. PubMed | Google Scholar
- Kong J, Kim SR, Binley K et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311-20. PubMed | Google Scholar
- Mathis T, de Bats F, Abouaf L, Kodjikian L. Dystrophie maculaire de stargardt. Images en Ophtalmologie. Mars-avril 2014; Vol VIII(numéro 2). PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures



Article metrics
PlumX Metrics
Dystrophie maculaire de Stargardt - ŕ propos d’un casRecently from the PAMJ








Authors´ services
