
Diagnostic tardif d’une hyperoxalurie primitive au stade d’insuffisance rénale chronique terminale avec hypoparathyroďdie sévčre
Zineb El Ghali, Zineb Ait Lahcen, Wafaa Fadili, Abderrahim Idrissi Kaitouni, Mohamed Hakkou, Abderrachid Hamdaoui, Inass Laouad
Corresponding author: Zineb El Ghali, Service de néphrologie – hémodialyse – transplantation rénale, hôpital Ibn Tofeil, CHU Mohammed VI, rue el mostachfa, Gueliz, 40000, Marrakech, Maroc. 
Received: 08 Jul 2013 - Accepted: 19 Mar 2014 - Published: 18 Apr 2014
Domain: Clinical medicine
Keywords: Hyperoxalurie, insuffisance rénale chronique terminale, douleurs osseuses, anémie
©Zineb El Ghali et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Zineb El Ghali et al. Diagnostic tardif d’une hyperoxalurie primitive au stade d’insuffisance rénale chronique terminale avec hypoparathyroďdie sévčre. Pan African Medical Journal. 2014;17:297. [doi: 10.11604/pamj.2014.17.297.3076]
Available online at: https://www.panafrican-med-journal.com//content/article/17/297/full
Original article 

Diagnostic tardif d’une hyperoxalurie primitive au stade d’insuffisance rénale chronique terminale avec hypoparathyroďdie sévčre
Diagnostic tardif d’une hyperoxalurie primitive au stade d’insuffisance rénale chronique terminale avec hypoparathyroďdie sévčre
Zineb El Ghali1,&, Zineb Ait Lahcen1, Wafaa Fadili1, Abderrahim Idrissi Kaitouni2, Mohamed Hakkou3, Abderrachid Hamdaoui3, Inass Laouad1
1Service de néphrologie – hémodialyse – transplantation rénale, hôpital ibn Tofeil, CHU Mohammed VI, Marrakech, Maroc, 2Centre d’hémodialyse, secteur libéral, Maroc, 3Laboratoire d’anatomopathologie, secteur libéral, Maroc
&Auteur correspondant
Zineb El Ghali, Service de néphrologie – hémodialyse – transplantation rénale, hôpital Ibn Tofeil, CHU Mohammed VI, rue el mostachfa, Gueliz, 40000, Marrakech, Maroc.
L’hyperoxalurie primitive est une anomalie métabolique congénitale rare caractérisée par un excčs de production avec accumulation d’oxalate secondaire ŕ un déficit enzymatique hépatique. Nous rapportons le cas d’un patient de 18 ans qui avait comme antécédent des lithiases rénales récidivantes depuis l’enfance évoluant vers l’insuffisance rénale chronique terminale, mis en hémodialyse périodique depuis 4 ans avec apparition d’une anémie résistante ŕ l’érythropoďétine pour laquelle il a été multitransfusé et qui était admis pour prise en charge de douleurs osseuses invalidantes d’aggravation progressive associées ŕ un syndrome tumoral fait d’adénopathies périphériques et de splénomégalie avec dyspnée et altération de l’état général. Le bilan réalisé avait objectivé de multiples images lytiques lacunaires et condensantes au niveau des poignets et des mains avec des petits reins calcifiés, une pleurésie avec péricardite de grande abondance drainée, une ascite de moyenne abondance avec splénomégalie homogčne, une anémie normochrome normocytaire ŕ 7.2 g/dl avec hyperferritinémie ŕ 1129 µg/l et un syndrome inflammatoire biologique. La calcémie était spontanément normale ŕ 97 mg/l avec hyperphosphorémie ŕ 83 mg/l et hypoparathyroďdie ŕ 74,85 pg/ml. Les PAL étaient ŕ 136 UI/l, l’aluminium sérique ŕ 13µg/l et la Vitamine D native ŕ 20,87ng/ml. Le diagnostic d’hyperoxalurie a été retenu sur les données de la biopsie ostéomédullaire objectivant des dépôts de cristaux d’oxalate de calcium avec fibrose médullaire et réaction macrophagique ŕ corps étranger. L’évolution a été marquée par la survenue de fractures spontanées récidivantes au niveau de l’épaule et des 2 hanches.
L’hyperoxalurie primitive est une anomalie congénitale du métabolisme responsable d’une surproduction endogčne d’oxalate liée ŕ un déficit enzymatique hépatique. C’est une maladie autosomique récessive rare dont les manifestations cliniques sont les conséquences de dépôt de cristaux d’oxalate de calcium insolubles en excčs au niveau du rein et des tissus. Le diagnostic est souvent posé ŕ un stade avancé avec des conséquences fatales.
Nous rapportons le cas d’un jeune garçon de 18 ans, 3čme d’une fratrie de 5, issu d’un mariage non consanguin, référé d’un centre d’hémodialyse privé pour prise en charge de douleurs osseuses avec altération de l’état général et syndrome tumoral. Le patient avait un long passé de lithiases rénales récidivantes depuis l’âge de 6 ans traitées par lithotripsie ŕ plusieurs reprises sans bilan étiologique. Il a été mis en hémodialyse en 2008 pour IRCT sur néphropathie interstitielle d’origine lithiasique. L’évolution en hémodialyse a été marquée par l’apparition d’une anémie résistante ŕ l’érythropoďétine pour laquelle il a été multitransfusé. 6 mois avant son admission, le malade avait présenté des douleurs osseuses des membres et du rachis d’installation progressive avec arthralgies inflammatoires des épaules, coudes, poignets et genoux. La symptomatologie s’est aggravée 2 mois avant l’hospitalisation par accentuation des douleurs devenues permanentes, aggravées lors des séances d’hémodialyse, entrainant une impotence fonctionnelle avec apparition de douleurs thoraciques et d’une dyspnée de repos sans toux ni hémoptysie évoluant dans un contexte de sensations fébriles et d’altération profonde de l’état général. Un traitement antibacillaire d’épreuve avait été démarré par le néphrologue référant aprčs bilan radiologique pour tuberculose pleuropulmonaire arręté aprčs un mois devant la non amélioration.
A l’admission, le patient était conscient, altéré, pâle, fébrile ŕ 38°C, tachycarde ŕ 140 battements/min, hypertendu ŕ 160/90 mmHg avec des oedčmes blancs des 2 membres inférieurs. L’examen ostéoarticulaire avait objectivé une douleur ŕ la palpation des différents segments osseux au niveau des membres, côtes et épineuses rachidiennes. L’examen cardiovasculaire avait mis en évidence une diminution des bruits du cśur ŕ l’auscultation et l’examen pleuropulmonaire un syndrome d’épanchement liquidien basithoracique gauche. Une splénomégalie homogčne a été retrouvée ŕ l’examen abdominal avec une matité ŕ la percussion et l’examen des aires ganglionnaires avait mis en évidence une adénopathie axillaire gauche de 2cm/1cm ferme mobile avec de multiples ganglions lenticulaires au niveau jugulaire et inguinal.
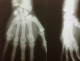
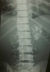
Le bilan radiologique avait objectivé de multiples images lytiques lacunaires et condensantes au niveau des poignets et des mains (Figure 1) avec une cardiomégalie associée ŕ une pleurésie et des petits reins calcifiés ŕ l’AUSP (Figure 2). L’échographie et la TDM abdominales avaient mis en évidence une ascite de moyenne abondance avec splénomégalie homogčne sans adénopathies profondes. L’échocardiographie avait retrouvé une hypertrophie ventriculaire gauche concentrique avec hypokinésie septale, dysfonction légčre du VG (fraction d’éjection ŕ 43%), forte HTAp (72 mmHg) et épanchement péricardique circonférentiel avec invagination de l’oreillette droite ce qui avait nécessité un drainage chirurgical en urgence qui avait ramené 1 litre de liquide jaune citrin transsudatif ŕ l’étude chimique.
Sur le plan biologique, notre malade avait une anémie normochrome normocytaire ŕ 7.2 g/dl, avec frottis sans particularité. La ferritine était ŕ 1129 µg/l et les LDH ŕ 426 UI/L. Cette anémie était résistante ŕ l’érythropoďétine ce qui avait nécessité des transfusions multiples, avec un syndrome inflammatoire: VS ŕ 130mm et augmentation des alpha 2 globulines ŕ l’électrophorčse des protéines sériques. Le bilan phosphocalcique avait objectivé une calcémie spontanément normale ŕ 97 mg/l avec hyperphosphorémie ŕ 83 mg/l et hypoparathyroďdie ŕ 74,85 pg/ml. Les PAL étaient ŕ 136 UI/L, l’aluminium sérique ŕ 13µg/l et la Vitamine D native ŕ 20,87 ng/ml.
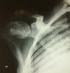
Une biopsie ganglionnaire a été faite au niveau axillaire et qui avait objectivé une adénite chronique réactionnelle, la biopsie péricardique avait mis en évidence un péricarde dystrophique sans signes de malignité et la biopsie ostéomédullaire était en faveur d’une hyperoxalurie avec dépôts de cristaux d’oxalate de calcium, fibrose médullaire et réaction macrophagique ŕ corps étranger (Figure 3, Figure 4). L’évolution a été marquée par la survenue de fractures spontanées ŕ répétition au niveau de l’épaule (Figure 5) et des 2 hanches traitées d’un côté par prothčse.
L’hyperoxalurie primitive est une pathologie rare dont l’incidence est estimée ŕ moins de1 cas / million d’habitants/ an [1]. Il s’agit d’une anomalie congénitale du métabolisme hépatique entrainant une surproduction d’oxalate avec excčs de son élimination urinaire. 3 formes ont été décrites dans la littérature correspondant chacune ŕ un déficit enzymatique particulier mais toutes les trois de transmission autosomique récessive. L’hyperoxalurie primitive de type 1 étant la forme la plus fréquente [2].
La présentation clinique chez notre patient était semblable aux données de la littérature: lithiases urinaires récidivantes depuis le jeune âge avec néphrocalcinose [3-5]. Cependant, le diagnostic a été posé tardivement au stade d’IRCT aprčs 4 ans de mise en hémodialyse avec oxalose systémique.
Les douleurs osseuses invalidantes aggravées par l’ultrafiltration et donc limitant cette derničre ce qui a aboutit ŕ la surcharge (HTA, śdčmes, ascite et pleuropéricardite) étaient le principal signe d’appel, associées ŕ un syndrome tumoral fait de splénomégalie et d’adénopathies avec altération profonde de l’état général ce qui avait motivé la réalisation de multiples biopsies (péricardique, ganglionnaire et ostéomedullaire) en vu d’éliminer une tuberculose ou une hémopathie maligne et c’était la biopsie ostéomédullaire qui avait permis de confirmer le diagnostic de la néphropathie initiale de notre patient aprčs 4 ans de mise en hémodialyse en objectivant les dépôts de cristaux d’oxalate au niveau osseux avec fibrose médullaire expliquant l’anémie résistante ŕ l’érythropoďétine. Les lésions osseuses observées au cours de l’oxalose sont liées ŕ la réaction inflammatoire induite par les cristaux d´oxalate qui provoque la résorption osseuse. Des fractures peuvent se produire spontanément dans ces zones de faiblesse [6]. Certains signes radiologiques sont caractéristiques de l’oxalose: bandes métaphysaires denses, ostéocondensation des vertčbres. Cependant, il est en pratique difficile de faire la part entre les lésions liées ŕ l’oxalose et celles induites par l’hyperparathyroďdie chez les malades en IRCT. La résorption osseuse semble ętre le résultat des deux [6, 7]. Notre patient avait par contre une hypoparathyroďdie qui semble ętre le facteur aggravant des lésions osseuses. Un cas similaire d’hypoparathyroďdie sur oxalose systémique a été rapporté chez un patient de 47 ans chez qui le diagnostic d’hyperoxalurie a été posé aprčs 6 ans de mise en hémodialyse [8].
La tendance spontanée ŕ la normocalcémie observée dans notre cas serait liée ŕ la stimulation de l’activité ostéoclastique par les macrophages constituant les granulomes.
Sur le plan thérapeutique, théoriquement, plusieurs mesures sont proposées: hyperhydratation mais qui reste difficile au stade d’insuffisance rénale chronique vu le risque de surcharge hydrosodée et d’śdčme aigu du poumon, inhibiteurs de cristallisation, administration de pyridoxine, mais la double transplantation hépatorénale reste le traitement de choix notamment au stade d’insuffisance rénale terminale [2]. Cette option thérapeutique n’était pas possible dans notre contexte.
Notre observation illustre les conséquences fatales liées au retard du diagnostic de l’oxalurie primitive en l’absence d’une possibilité de transplantation hépatorénale qui reste le seul traitement efficace de cette pathologie.
Les auteurs ne déclarent aucun conflit d'intérêt.
Tous les auteurs ont également contribué à la conduite de ce travail et à la rédaction de cet article. Tous les auteurs ont lu et approuvé la version finale du manuscrit.
Figure 1: Radiographie des mains prenant les poignets: multiples images lytiques et condensantes
Figure 2: AUSP image de néphrocalcinose bilatérale avec réduits de taille
Figure 3: Biopsie ostéomédullaire:
cristaux d’oxalate de calcium déposés en rosettes ou en étoiles
Figure 4: Biopsie ostéomédullaire, fort grossissement; Dépôts de cristaux d’oxalate de calcium avec réaction macrophagique ŕ corps étranger
Figure 5: Radiographie
de l’épaule; déminéralisation diffuse avec fracture pathologique du col huméral
- Cochat Pierre et al. Epidemiology of primary hyperoxaluria type Nephrol Dial Transplant. 1995; 10(Suppl 8):3-7.PubMed | Google Scholar
- Harambat Jérôme, Fargue Sonia, Bacchetta Justine, Acquaviva Cécile, Cochat Pierre. Primary Hyperoxaluria. Int J Nephrol. 2011; 2011:864580. PubMed | Google Scholar
- Gargah Tahar, Khelil Nourchčne, Youssef Gharbi, Karoui Wiem, Lakhoua Mohamed Rachid, Abdelmoula Jaouida. primary hyperoxaluria type 1 in tunisien children. Saudi J Kidney Dis Transpl. Mar 2012; 23(2):385-90. PubMed | Google Scholar
- Martín Mickael, Martín Reyes Guillermo, Torres de Rueda Alvaro. Delayed diagnosis of primary hyperoxaluria in a young patient with advanced chronic renal failure. Nefrologia. 2011; 31(2):227-9. PubMed | Google Scholar
- Bogle Mary Anna et al. Primary hyperoxaluria in a 27-year-old woman. J Am Acad Dermatol. Oct 2003; 49(4):725-8. PubMed | Google Scholar
- El Hage Samer, Ghanem Ismat, Baradhi André, Mourani Chebel, Mallat Samir, Dagher Fernend, Kharrat Khalil. Skeletal features of primary hyperoxaluria type 1, revisited. J Child Orthop. Jun 2008; 2(3):205-10.. PubMed | Google Scholar
- Brancaccio Diego, Poggi Alessandro, Ciccarelli Carmela, Bellini Federica et al. Bone changes in end stage oxalosis. AJR Am J Roentgenol. May 1981; 136(5):935-9. PubMed | Google Scholar
- Karadag Serhat, Gursu Meltem, Aydin Zeki, Uzun Sami, Dogan Ozdemir, Ozturk Savas. Primary hyperoxaluria in an adult presenting with end-stage renal failure together with hypercalcemia and hypothyroidism. Hemodial Int. Oct 2011; 15(4):573-6. PubMed | Google Scholar
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
Recently from the PAMJ








Authors´ services
