
Dermatomyosite du sujet âgé: étude de 4 observations dans le sud tunisien
Faten Frikha, Mouna Snoussi, Raida Ben Salah, Noura Saidi, Neila Kaddour, Zouhir Bahloul
Corresponding author: Faten Frikha, Service de Médecine interne CHU Hédi Chaker 3029 Sfax-Tunisie 
Received: 23 Dec 2011 - Accepted: 02 Jun 2012 - Published: 06 Oct 2012
Domain: Clinical medicine
Keywords: Myopathie inflammatoire, dermatomyosite, personnes âgées
©Faten Frikha et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Faten Frikha et al. Dermatomyosite du sujet âgé: étude de 4 observations dans le sud tunisien. Pan African Medical Journal. 2012;13:26. [doi: 10.11604/pamj.2012.13.26.1451]
Available online at: https://www.panafrican-med-journal.com//content/article/13/26/full
Original article 

Dermatomyosite du sujet âgé: étude de 4 observations dans le sud tunisien
Dermatomyosite du sujet âgé: étude de 4 observations dans le sud tunisien
Faten Frikha1,&, Mouna Snoussi1, Raida Ben Salah1, Noura Saidi, Neila Kaddour1, Zouhir Bahloul1
1Service de Médecine interne CHU Hédi Chaker 3029 Sfax, Tunisia
&Auteur correspondant
Faten Frikha, Service de Médecine interne CHU Hédi Chaker 3029 Sfax-Tunisie
Les myopathies inflammatoires (MI) sont des connectivites rares caractérisées par l’association d’un déficit musculaire et la présence d’un infiltrat inflammatoire au sein du muscle squelettique. Les MI regroupent trois entités principales : la polymyosite (PM), la dermatomyosite (DM) et la myosite ŕ inclusions [1,2]. Elles sont dotées d’un grand polymorphisme clinique et évolutif. Des désordres immunitaires cellulaires et humoraux sont probablement ŕ l’origine de ces connectivites. Des progrčs considérables ont été réalisés ces derničres années dans la compréhension de leur pathogénie. La DM se distingue par une atteinte cutanée caractéristique. Elle est caractérisée par une atteinte primitive péri vasculaire de mécanisme humoral [2,3]. Le diagnostic de cette affection repose sur un faisceau d’arguments cliniques, enzymologiques, électromyographiques et histologiques. La DM touche essentiellement l’adolescent et l’adulte jeune en Tunisie [4], elle est trčs rare chez le sujet âgé, le plus souvent associée ŕ une pathologie cancéreuse [5]. Le traitement et le pronostic restent comparables. Le but de travail est de déterminer, ŕ travers une étude rétrospective, les caractéristiques épidémiologiques, cliniques, para cliniques, évolutives et les modalités thérapeutiques de quatre patients dont l’âge était supérieur ou égal ŕ 65 ans au moment du diagnostic de la myosite (dermatomyosite) parmi une cohorte de 78 patients atteints de MI (44 cas atteints de DM) , suivis au service de médecine interne du CHU Hčdi Chaker de Sfax entre 1979 et 2010 et de les comparer ŕ une population témoin plus jeune.
Nous rapportons une étude rétrospective incluant 4 patients dont l’âge était supérieur ŕ 65 ans au moment du diagnostic de la DM parmi une cohorte de 44 patients atteints de DM, suivis au service de médecine interne du CHU Hčdi Chaker de Sfax durant une période de 31 ans s’étalant de Janvier 1979 ŕ décembre 2010. Le diagnostic de DM a été retenu en se basant sur les critčres de Bohan et Peter [1].
Nous avons étudié les données épidémiologiques (âge, sexe), le mode de début (aigu, subaigu, chronique), le délai du diagnostic ( durée qui sépare la date d’apparition du premier signe de la maladie de la date du diagnostic définitif), les signes cliniques en rapport avec la maladie (signes généraux, cutanés, musculaires, digestifs, pulmonaires, cardiaques, neurologiques et rénaux), les données biologiques ( recherche d’un syndrome inflammatoire, dosage des enzymes musculaires, étude immunologique), l’étude des données de l’électromyogramme (EMG) ŕ la recherche d’un syndrome myogčne et l’étude anatomopathologique (biopsie musculaire et cutanée). Nous avons précisé les modalités thérapeutiques (en précisant les molécules utilisées, leur posologie, la durée du traitement) ; ainsi que les données évolutives (en se basant sur les éléments cliniques et biologiques).
Observation 1
Mr A.A. âgé de 84 ans, aux antécédents d’insuffisance cardiaque, était hospitalisé en novembre 2001 pour arthralgies de type inflammatoire touchant les grosses articulations (hanches, genoux et épaules) associées ŕ des troubles de la déglutition avec une dysphagie, des fausses routes itératives évoluant depuis deux mois motivant son hospitalisation. L’examen clinique objectivait un érythčme sans śdčme siégeant au niveau du visage en périorbitaire et au niveau du décolleté, un déficit musculaire des ceintures scapulaires et pelviennes au testing musculaire. L’examen neurologique révélait des réflexes ostéo-tendineux faibles. Le reste de l’examen clinique était sans particularité. Le bilan biologique montrait un syndrome inflammatoire biologique avec une vitesse de sédimentation (VS) accélérée ŕ 80 mm la premičre heure, un taux de CRP élevé ŕ 92 mg/l, une hypo albuminémie ŕ 30g/l, une élévation des alpha 1 globulines (3g/l), alpha 2 globulines (10,2g/l) et gamma globulines (15g/l). On notait également une insuffisance rénale avec une urée ŕ 23 mmol/l et une créatinine ŕ 212 umol/l. L’hémogramme retrouvait un taux de leucocytes ŕ 12000 éléments/mm3 avec une hémoglobine ŕ 12,9 g/dl et des plaquettes normales. On notait également une élévation modérée des transaminases avec un taux des ASAT et des ALAT ŕ deux fois la normale, des CPK ŕ 1380 UI/l et des LDH ŕ 1110 UI/l. La recherche d’anticorps anti-nucléaires était positive ŕ 1/80 de type hépatites B et C était négatives. La biopsie musculaire du deltoďde confirmait l’existence d’une dematopolymyosite (DPM) aiguë en montrant des signes histologiques de myopathie inflammatoire avec un infiltrat inflammatoire lympho-histiocytaire dans les régions septales, une inégalité de la taille des fibres des fibres atrophiques arrondies, des fibres en voie de nécrose, absence d’atrophie péri fasciculaire. L’électromyogramme (EMG) montrait des signes d’atteinte neurogčne périphérique démyélinisante proximale et distale sensitivomotrice pouvant ętre en rapport avec une forme sévčre de sa DM. Devant ces données, le diagnostic de Dermatomyosite (DM) était retenu selon les critčres de Bohan et Peter [1]. L’enquęte ŕ la recherche d’une néoplasie profonde associée était négative avec un examen ORL normal, des marqueurs tumoraux (AFP, ACE, CA 19-9) normaux, une radiographie thoracique et une échographie abdominopelvienne normales. Le patient était traité par trois bolus de méthylprednisolone (solumédrol®) ŕ la dose de 1 g/jour relayés par une corticothérapie ŕ forte dose (prednisolone 1mg/kg/j soit Solupred® 3 comprimés par jour) avec une amélioration partielle des forces musculaires et des fausses routes et du taux des enzymes musculaires (CPK ŕ 144 UI/l et LDH ŕ 707 UI/l).
Observation 2
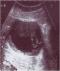
Madame K.H. âgée de 86 ans, aux antécédents d’arythmie complčte par fibrillation auriculaire depuis 9 ans sous Cordarone et Sintrom, hypothyroďdie sous L-thyroxine, était hospitalisée en juin 2008 pour des myalgies diffuses avec déficit musculaire des deux ceintures, śdčme et érythčme du visage évoluant depuis trois mois. A l’admission, la patiente était apyrétique, pesait 64 Kg et sa Pression artérielle mesurée ŕ 120/70 mmHg. L’examen clinique trouvait un érythčme du visage et en périorbitaire en lunette, des lésions érythémato-squameuses au niveau du décolleté. On objectivait un déficit musculaire des deux ceintures scapulaires et pelviennes avec un score ŕ 63/75 sur l’échelle fonctionnelle des myosites. La biologie montrait une anémie ŕ 9 g/dl hypochrome microcytaire, des leucocytes ŕ 20000 éléments/mm 3, des plaquettes normales, une vitesse de sédimentation ŕ 38 mm la premičre heure, un taux de CRP ŕ 29 mg/l. on notait également une insuffisance rénale avec un taux d’urée ŕ 13,3 mmol/l et une créatinine ŕ 178 umol/l. Les CPK étaient franchement élevées ŕ 7504 UI/l/. Les transaminases étaient élevées avec un taux d’ASAT et d’ALAT ŕ 387 et 292 UI/l respectivement. La recherche d’anticorps anti-nucléaires était positive ŕ 1/1280 de type homogčne. Le bilan thyroďdien était normal. L’électromyogramme (EMG) montrait des signes d’atteinte myogčne diffuse avec des potentiels de fibrillation caractéristique d’une polymyosite en phase active. Le diagnostic d’une dermatomyosite DM était alors retenu. Devant l’âge avancé, l’origine secondaire de sa maladie était évoquée. Un bilan ŕ la recherche d’une néoplasie profonde trouvait un taux de CA 125 élevé ŕ 95,1 UI/ml (la valeur normale étant inférieure ŕ 35UI/ml). L’échographie abdominopelvienne montrait une deux masses kystiques sus et latéro-utérine ŕ paroi épaissie renfermant des bourgeonnements tissulaires (Figure 1). Ces masses étaient d’origine vraisemblablement ovarienne mesurant respectivement 5X8X10 cm et 4,8X6,6 cm. Le diagnostic de DM secondaire ŕ une néoplasie ovarienne était alors retenu.
La radiographie thoracique objectivait une cardiomégalie avec une image calcifiée du champ pulmonaire gauche séquellaire sans signe de malignité. Devant le déficit musculaire important et la myolyse sévčre avec insuffisance rénale, la patiente était mise sous prednisone ŕ forte dose (1mg/kg/j). L’évolution était marquée par l’amélioration partielle de la myolyse avec un taux de CPK ŕ 3280 UI/l, l’absence d’amélioration du déficit musculaire, l’aggravation de son état respiratoire avec l’installation d’une insuffisance respiratoire aiguë nécessitant son transfert en réanimation, décision que les parents avaient refusé, la patiente était alors mise sortante contre avis médical.
Observation 3
Madame B.M. âgée de 71 ans, aux antécédents de fracture de la hanche gauche avec mise en place d’une prothčse, était hospitalisée dans le service en Aoűt 2003 pour oedčme du visage, des membres supérieures et un érythčme orbitaire, le tout évoluant de puis quinze jours. L’examen clinique objectivait des lésions érythémato-maculeuses mal limitées siégeant au niveau des pommettes et des paupičres avec śdčme ŕ ce niveau, de la face antérieure des deux genoux et jambes. Le reste de l’examen clinique était sans particularité notamment l’absence de déficit musculaire des ceintures avec un signe du tabouret négatif. La patiente signalait la notion d’un acrosyndrome des mais avec ŕ la capillaroscopie un aspect de microangiopathie organique et présence de mégacapillaires. A la biologie, on trouvait une hémoglobine ŕ 11,6 g/dl, des leucocytes et des plaquettes normales. La vitesse de sédimentation était accélérée ŕ 67 mm la premičre heure, le dosage de la CRP était négatif, les transaminases étaient également normales, les anticorps anti-nucléaires et les anticorps anti-phospholipides étaient négatifs. Le taux des CPK était élevé ŕ 665 UI/l, les LDH étaient ŕ 764 UI/l et les aldolases ŕ 8,22 UI/l. Le diagnostic de DPM était alors évoqué. Un EMG pratiqué concluait ŕ un tracé myogčne riche et peu ample au niveau des deltoďdes compatibles avec une DPM peu sévčre. La biopsie musculaire montrait la présence de quelques foyers inflammatoires au niveau des septa inter fasciculaire et dans l’endomysium évoquant une myopathie inflammatoire. Un bilan ŕ la recherche d’une néoplasie profonde pratiqué concluait ŕ un dosage normal des marqueurs tumoraux, un examen ORL sans anomalies, une radiographie thoracique montrant un syndrome interstitiel pouvant évoquer une atteinte pulmonaire dans le cadre de sa maladie, une échographie abdominopelvienne, échographie mammaire et fibroscopie oeso-gastro-duodénale étaient normales. L’électrophorčse des protéines sériques montrait la présence d’une fraction anormale migrant en zone gamma confirmée par l’immunoélectrophorčse qui objectivait la présence de chaînes légčres kappa et lambda dans le sang et dans les urines liés ŕ des taux élevés d’immunoglobulines de type Ig G (16,82 g/l) avec un taux subnormal des autres classes d’immunoglobulines. Le myélogramme ainsi que le bilan radiologique osseux étaient normaux.Le diagnostic d’une DM associée ŕ une gammapathie monoclonale bénigne était retenu. La patiente était mise sous corticothérapie ŕ forte dose : prednisone ŕ la dose de 1mg/kg/j pendant six semaines avec dégression progressive de 5 mg ŕ chaque semaine jusqu’ŕ une dose d’entretien de 10 mg/j. L’évolution était favorable pour sa DM avec marquée par amélioration progressive des lésions érythémato-oedémateuses et normalisation des enzymes musculaires. Cependant, la patiente développait un diabčte cortico-induit traité par des anti-diabétiques oraux. Elle avait un suivi régulier ŕ la consultation externe durant trois années successives et puis perdu de vue avec un recul évolutif de 39 mois.
Observation 4
Madame D.T âgée de 73 ans, sans antécédents pathologiques notables, était hospitalisée en Octobre 2008 pour l’exploration de lésions érythémateuses touchant le visage notamment au niveau des pommettes, le cuir chevelu, la face antérieure du thorax et la face antérieure des deux genoux évoluant depuis deux mois, compliqué un mois avant son admission par l’apparition de myalgies diffuses , de dysphagie avec modification de la voie devenant nasonnée, fausses routes ŕ répétition le tout évoluant dans un contexte d’altération de l’état général ŕ type d’asthénie, amaigrissement chiffré ŕ huit kilogrammes en deux mois et quinze jours motivant ainsi son hospitalisation. L’examen clinique objectivait la présence d’un érythčme télangiectasique qui touche les pommettes, les paupičres, les mains notamment en péri- unguéal avec signe de la manicure, le décolleté, le cuir chevelu et la face antérieure des deux genoux. L’examen ostéo-articulaire mettait en évidence un déficit musculaire au niveau de la ceinture scapulaire avec difficulté ŕ la surélévation des deux membres supérieures au delŕ de 90 degré plus marqué du coté gauche. L’examen oto-rhino laryngé montrait une muqueuse buccale et oro-pharyngée pale, un voile du palais symétrique et mobile, le larynx et l’hypopharynx et le cavum étaient normaux. L’examen cardio-pulmonaire, neurologique, ganglionnaire, digestif et génital était normal.
A la biologie, on notait un taux de leucocytes normal ŕ 9900 éléments/mm3, une hémoglobine ŕ 12,9g/dl, des plaquettes ŕ 255000 éléments/mm3, la vitesse de sédimentation était ŕ 25 mm la premičre heure, l’électrophorčse des protéines sériques était normale, le bilan rénal était également sans anomalie. Le dosage des transaminases montrait des ASAT ŕ 71UI/l, les ALAT était ŕ 58UI/l, les CPK étaient ŕ 827 UI/l, les LDH ŕ 809UI/l. Le bilan immunologique concluait ŕ un taux d’anticorps anti-nucléaires positif ŕ un taux de 1/640. Les anticorps anti-cytoplasme des polynucléaires neutrophiles étaient négatifs ainsi que les anticorps anti-phospholipides. Devant ces données cliniques et biologiques, le diagnostic de dermatopolymyosite a été évoqué. La patiente avait bénéficié d’un EMG des quatre membres qui s’est révélé normal. Une biopsie cutanéo-muqueuse était faite devant l’aggravation des lésions érythémateuses concluant ŕ un épiderme aminci avec atrophie du corps muqueux, la membrane basale était pigmentée et rectiligne, le derme superficiel contenant un discret infiltrat lympho-histiocytaire, le réseau élastique était focalement détruit. La biopsie neuromusculaire pratiquée au niveau du muscle deltoďde montrait une discrčte inégalité des fibres avec présence de fibres nécrotiques ŕ cytoplasme pâle, d’autres sont en voie de résorption macrophagique, les coupes sériées en paraffine montraient des infiltrats inflammatoires lymphoplasmocytaires au niveau des septas et des régions péri vasculaire, il n’avait pas d’atrophie péri fasciculaire, cet aspect histologique était compatible avec le diagnostic d’une DPM. Devant l’altération profonde de l’état général et l’âge avancé, un bilan ŕ la recherche d’une néoplasie profonde était réalisé concluant ŕ des marqueurs tumoraux négatifs, la radiographie thoracique, l’échomammographie, l’échographie abdominopelvienne, la fibroscopie digestive, et l’échographie cervicale étaient sans anomalies. Devant la présence de signe de gravité clinique évoquant une atteinte des muscles laryngo-pharyngés avec dysphagie importante, dysphonie, fausses routes et l’apparition d’un reflux de liquide par le nez, la patiente était traitée par trois bolus de méthylprednisolone relayée par une corticothérapie ŕ forte dose 1mg/kg/jour pendant six semaines avec dégression progressive et maintient d’une dose d’entretient de 20 mg/j de prednisone, cette corticothérapie était associé ŕ un traitement immunosuppresseur : méthotrexate ŕ la dose de 15mg/semaine. L’évolution était marquée par l’aggravation initiale de l’atteinte pharyngée avec dysphagie importante une semaine aprčs le démarrage de la corticothérapie nécessitant la mise en place d’une sonde naso-gastrique durant un mois. On notait également une amélioration progressive des lésions cutanées et des forces musculaires avec normalisation du taux des CPK et LDH. La corticothérapie était compliquée chez cette patiente par l’apparition d’une ostéoporose confirmée par ostéodensitométrie.
Le recul évolutif est de trente quatre mois. La patiente est encore suivie ŕ la consultation externe, le dernier contrôle clinique montrait quelques lésions érythémato-squameuses du visage et du décolleté, absence de déficit moteur des ceintures scapulaires et pelviennes, le taux des CPK était normal (188 UI/l), les LDH étaient légčrement élevées ŕ 577 UI/l.
Etude descriptive
Parmi les 78 cas de myopathies inflammatoires colligés dans le service durant la période d’étude, 4 patients avaient un âge supérieur ou égal ŕ 65 ans au moment du diagnostic de la maladie, soit une fréquence de 5,12 % de toutes les myosites. Il s’agissait d’une dermatomyosite (DM) dans les 4 cas soit une fréquence de 9,09% des DM diagnostiquées (4 parmi 44 cas de DM). L’âge moyen du début de la DM était de 78,5 ans avec des extręmes allant de 71 ŕ 86 ans. Il s’agissait de 3 femmes et un homme. Le sexe ratio H/F était de 0, 33. La comparaison avec le groupe de DM dont l’âge est < 65 ans ne trouve pas différence significative pour le sexe (p=0,910). Le délai moyen du diagnostic de la DM était de 2 mois avec des extręmes allant de 15 jours ŕ 3 mois. Les signes cutanés (érythčme et/ou śdčme du visage) étaient les plus fréquents retrouvés dans 75 % des cas. Les signes généraux étaient présents chez un patient. Il s’agissait d’une asthénie associée ŕ un amaigrissement. L’atteinte musculaire initiale était fréquente (50%). Elle était absente dans les deux autres cas. Les myalgies étaient présentes chez deux patients (50%). Le déficit musculaire proximal était noté ŕ l’examen clinique dans trois cas (75%). Il a intéressé la ceinture scapulaire et pelvienne. L’atteinte des muscles laryngo-pharyngés était fréquente (50%). L’atteinte cutanée était dominée par l’érythčme périorbitaire noté dans 4 cas associé ŕ un śdčme dans 2 cas. Les autres localisations de l’érythčme étaient : péri unguéal dans un cas avec signe de la manicure, décolleté dans 3 cas, faces d’extension des genoux dans 2 cas et cuir chevelu dans un cas. Le syndrome de Raynaud était présent dans un cas, la capillaroscopie objectivait un aspect de microangiopathie organique et présence de mégacapillaires. Des arthralgies inflammatoires des grosses articulations étaient signalées dans un seul cas, elles touchaient les hanches, les genoux et les épaules. Les manifestations digestives étaient notées dans 2 cas ŕ type de dysphagie haute avec fausses routes. Elles étaient inaugurales de la maladie dans les 2 cas. L’atteinte pulmonaire était notée dans un seul cas ŕ type de détresse respiratoire survenue au cours de l’évolution de la maladie.
Un syndrome inflammatoire biologique (SIB) était présent chez 3 patients. La valeur moyenne de la vitesse de sédimentation chez nos 4 malades était de 52,5 mm ŕ la premičre heure (38 ŕ 80 mm H1).Les enzymes musculaires étaient élevées chez tous nos patients. L’augmentation des CPK était notée dans 4 cas, celle de la LDH dans trois cas. Les transaminases étaient élevées chez 3 patients. Les anticorps antinucléaires étaient recherchés chez tous les patients. Ils étaient positifs chez trois patients.
L’électromyogramme (EMG) était pratiqué dans tous les cas. Il montrait un tracé myositique dans 2 cas, neurogčne dans un cas, le tracé était normal dans l’autre cas. La biopsie musculaire était réalisée chez 3 patients. Elle était compatible avec une myosite certaine, l’inflammation et la nécrose étaient les aspects les plus fréquents. L’association ŕ une hypothyroďdie était notée dans un cas. La DM était associée ŕ une néoplasie ovarienne chez une patiente. Elle était d’apparition concomitante. Dans notre série, tous nos patients ont été traités. Les moyens thérapeutiques étaient basés sur les corticoďdes, les immunosuppresseurs et le traitement symptomatique. La corticothérapie était indiquée en premičre intention chez tous les patients et administrée ŕ forte dose (1mg/Kg/j) d’équivalent Prednisone. Elle était initiée par des bolus de méthylprednisolone ŕ raison de 1g/j trois jours de suite chez 2 patients. Ces bolus étaient indiqués devant la gravité du tableau clinique dans ces 2 cas avec atteinte des muscles laryngo-pharyngés, la dysphagie importante, la dysphonie, les fausses routes et l’apparition d’un reflux de liquide par le nez. Le recours au traitement immunosuppresseur était indiqué chez un seul patient. La molécule était le méthotrexate ŕ la dose de 15 mg/semaine. La durée moyenne du suivi était de 19 mois (2 - 39 mois). L’évolution était défavorable chez une patiente avec installation d’une insuffisance respiratoire aigue nécessitant sa sortie contre avis médical avec décčs immédiat. Un patient était perdu de vue aprčs sa sortie de l’hôpital. Les deux autres patients avaient un suivi plus prolongé respectivement de 34 et 39 mois avec une amélioration partielle de leur symptomatologie sous corticothérapie. La morbidité iatrogčne due ŕ l’utilisation des corticoďdes a concerné deux patients. Il s’agissait d’un diabčte cortico-induit nécessitant la mise du patient sous anti-diabétiques oraux et d’ostéoporose confirmée par ostéodensitométrie pour l’autre patient.
La comparaison entre les deux groupes DM du sujet jeune (âgé de moins de 65 ans) et DM du sujet âgé trouve les résultats suivants: Le délai diagnostique moyen de la DM du sujet âgé était de 2 mois versus 6,1 mois par rapport ŕ la myosite du sujet jeune ; le mode de début était essentiellement aigu (p=0,001) ; la néoplasie était plus fréquente mais sans différence significative (p=0,25) (Tableau 1).
La DM est une connectivite rare ; son incidence annuelle varie de 2 ŕ 10 cas/millions d’habitants selon les populations étudiées [2,6,7]. La disparité de l’incidence annuelle selon les pays serait expliquée par des facteurs génétiques et environnementaux [8]. En terme de fréquence tout âge confondu, la DM est la plus fréquente des myopathies inflammatoires notamment chez l’enfant et l’adulte jeune. La survenue de la DM aprčs l’âge de 65 ans est une éventualité rare [5]. La fréquence des myopathies inflammatoires du sujet âgé est difficile ŕ évaluer compte tenu de l’hétérogénéité des populations étudiées [9-11]. Peu d’études se sont intéressées sur les myopathies inflammatoires survenant ŕ un âge tardif [9,10]. Dans l’étude de Pautas et al [9], colligeant 200 cas de PM/DM, les auteurs trouvent 21 patients qui sont âgés de plus de 65 ans soit une fréquence de 10,5 %. Marie et al [10] rapportent une fréquence de 29 % (23 cas parmi une série de 79 patients atteints de PM/DM). Dans notre service parmi les 78 sujets suivis pour une myopathie inflammatoire, 4 patients (5,12%) avaient un âge > ŕ 65 ans soit une fréquence moindre que celle rapportée dans la littérature. La DM peut survenir ŕ n’importe quel âge mais elle touche essentiellement l’adulte [12]. Elle possčde deux pics de fréquence : entre 5 et 14 ans pour l’enfant et dans la 5čme ou 6čme décennie chez l’adulte [2]. Chez le sujet âgé, la DM reste rare et le plus souvent associée ŕ une pathologie néoplasique [5]. L’âge moyen de nos patients était de 78,5 ans avec des extręmes allant de 71 ŕ 86 ans versus un âge moyen de 35,4 + 15,4 ans (12-64) dans le groupe sujet jeune. Dans la série de Pautas [9], l’âge moyen des 21 patients âgés était de 69,9+ 4,8 ans (extręmes de 65 et 80 ans) versus un âge moyen de 46.4 ± 12.4 ans (extręmes de 23 - 64) pour les patients de moins de 65 ans avec des différences significatives entre les 2 groupes. La DM est plus fréquente chez les femmes que chez les hommes quel que soit l’âge mais cette différence semble s’atténuer chez les sujets les plus âgés.
Une prédominance féminine était constatée chez nos patients (sex ratio femme/homme ŕ 3), ces résultats ont été trouvés dans différentes séries de la littérature [4,5,13,14]. L’installation de la maladie est souvent insidieuse et progressive [15]. Il faut y penser devant des manifestations de faiblesse, de fatigue, d’intolérance ŕ l’effort et ne pas négliger ces symptômes chez les sujets âgés [15]. La DM du sujet âgé se caractérise par un retard diagnostique et une fréquence des formes chroniques [9]. Chez nos patients, le début était aigu avec un délai moyen de diagnostic de 2 mois. L’atteinte musculaire peut inaugurer la maladie, et elle est représentée essentiellement par le déficit musculaire et les myalgies [16]. Le déficit touche typiquement la musculature striée de façon bilatérale, symétrique et non sélective. Il prédomine sur les muscles proximaux notamment sur la ceinture scapulaire et surtout pelvienne et sur les muscles cervicaux. L’intensité de la faiblesse est variable d’u sujet ŕ l’autre [12]. Ce déficit était noté chez 75% de nos patients, sa fréquence varie selon la littérature de 60 ŕ 90% [1,4,17]. Les myalgies sont rarement associées au déficit musculaire et peuvent ętre au premier plan dans les formes aigues. Elles sont spontanées ou provoquées par la palpation. La fréquence des myalgies varie de 25 ŕ 70% selon les séries [18].Dans sa série, Pautas et al [9] trouve que les myalgies sont moins fréquentes chez le sujet âgé (47.5% versus 71.5%, p > 0.05). Dans notre série, les myalgies étaient rapportées dans 50% des cas versus 87,5% chez le sujet plus jeune sans différence significative. Les manifestations cutanées peuvent précéder la myosite. Il s’agit d’un érythro-śdčme photosensible, prédominant dans les zones découvertes. L’érythčme orbitaire en lunette est typique et pathognomonique de la maladie ; il touche les paupičres et peut s’étendre aux joues sans atteindre la racine du nez [4,5]. L’érythčme peut toucher également la région péri unguéale donnant ainsi le signe de la manucure évocateur de la maladie [5]. Il s’agit d’un érythčme congestif, rouge vif et douloureux ŕ la pression [12]. L’érythčme peut s’étendre également au décolleté, face d’extension des genoux, coudes et mains. Dans notre série, l’érythčme périorbitaire était noté chez tous nos patients associé ŕ un śdčme dans deux cas. L’atteinte digestive est due ŕ l’inflammation de la musculature striée et lisse des différents segments du tube digestif [19]. Les troubles oesopharyngés résultent de l’atteinte de la musculature striée du pharynx et de la partie supérieure de l’śsophage se traduisant par une dysphagie et des fausses routes [20]. Dans la plupart des séries, ainsi que pour nos patients, l’atteinte des muscles oro-pharyngés est précoce et concomitante ŕ l’atteinte des muscles proximaux [21]. L’atteinte oesophagienne est plus fréquente chez les sujets âgés [22].
L’association des MI (PM/DM) ŕ une néoplasie, tout âge confondu, n’est pas fortuite et ce lien entre les deux pathologies est désormais bien établie. Sa fréquence varie entre 6 et 47,8% des cas selon les études [4,9-11,23,24] (Tableau 2). Sigurgeirsson et al ont étudié le risque de cancer sur une cohorte de 788 patients dont 392 DM ; le risque relatif de développer un cancer dans le groupe DM était de 2,4 pour les hommes et de 3,04 pour les femmes. Dans cette population, 40% des décčs sont secondaires au cancer [25]. Les DM du sujet âgé sont fréquemment associées ŕ une néoplasie profonde ; l’âge apparait comme un facteur prédictif de survenue du cancer au cours de cette connectivite [4]. En effet, l’association ŕ une néoplasie est estimée selon les études ŕ 10 % avant l’âge 65 ans et peut atteindre jusqu’ŕ 50 % des cas aprčs 65 ans [26]. Cette association est plus fréquente au-delŕ de 45 ans [27]. Selon Stockton, le risque de cancer associé est significatif au cours des DM pour des âges situés entre 45 et 74 ans [28].
Le diagnostic de néoplasie est dans la majorité des cas concomitant ŕ celui de la myosite ou fait quelques mois plus tard [4]. Le risque est plus élevé au cours de la premičre année qui suit le diagnostic de la DM et persiste jusqu’ŕ la cinquičme année aprčs le diagnostic [4,28]. Sur le plan biologique, les CPK sont souvent plus élevés et les auto- anticorps plus souvent négatifs dans les DM associées ŕ un cancer que dans les DM isolées [27]. Le type de néoplasie associé aux DM varie selon l’étude et la région géographique du patient. Les cancers gynécologiques notamment le cancer de l’ovaire sont les plus fréquents chez les femmes [29] avec plusieurs cas rapportés dans la littérature sous forme de cas [30-32]. Chez les hommes, il s’agit plutôt de cancer du poumon, digestifs et de lymphomes [33-35]. Chez le sujet âgé et selon la littérature, il existe une nette prédominance du cancer du colon [10] et de l’adénocarcinome rectal [9]. Le Tableau 3 résume quelques observations de DM du sujet âgé associées ŕ une néoplasie rapportée dans la littérature [36-38].
Ainsi devant une DM du sujet de plus de 40 ans, s’impose un bilan carcinologique répété surtout dans les cinq premičres années qui suivent le diagnostic. Ce bilan englobe un examen clinque complet avec les touchers pelviens inclus et systématiquement une radiographie thoracique, une échographie abdomino-pelvienne, une mamaographie chez les femmes, les marqueurs tumoraux (PSA et CA125). Les autres investigations se discuterons en fonction des données de l’examen : un scanner thoraco-abomino-pelvien et/ou une colonoscopie, chez les patients ayant des facteurs prédictifs de néoplasie [4,29]. On rajoute ŕ ces recommandations, la pratique d’un examen ORL systématique chez un patient provenant d’un pays endémique pour les néoplasies du nasopharynx (Maghreb et Asie), ce d’autant que le pronostic de ces néoplasies semble bon aprčs traitement par la corticothérapie et radiothérapie [35].
Le syndrome inflammatoire biologique (SIB) est fréquent au cours des DM. La vitesse de sédimentation est augmentée de façon modérée chez 50 ŕ 60% des cas [3]. Elle n’est pas corrélée ŕ l’intensité de la maladie et elle n’intervient pas dans le diagnostic. L’anémie et la polynucléose sont également fréquentes [19]. Le SIB était présent chez 3 de nos patients (75% des cas). L’augmentation des enzymes musculaires témoignant de la nécrose musculaire constitue un critčre diagnostique [3]. La créatine kinase (CPK) représente l’enzyme musculaire la plus spécifique, elle renseigne sur une souffrance objective du tissu musculaire. Elle est informative si elle est réalisée selon les recommandations d’usage aprčs 24 ŕ 48 heures de repos physique, et significative si les valeurs sont ŕ plus de 2 ŕ 3 fois la normale [15]. Les CPK sont augmentée dans 75 ŕ 85% des cas, cependant un taux normal ne doit pas écarter le diagnostic [3]. Les autres enzymes musculaires sont : l’aldolase sérique qui peut ętre utile (elle est parfois la seule augmentée), les transaminases et la lacticodéshydrogčnase sont moins spécifiques [3,19]. Chez nos patients, on a noté une élévation des CPK dans tous les cas, les LDH et les transaminases étaient élevées dans trois cas, l’aldolase était élevée chez un patient. Concernant les anomalies immunologiques, la fréquence des anticorps antinucléaires est estimée entre 30 ŕ 50% des cas dans les séries [3]. Dans notre série, les AAN étaient positifs chez trois de nos patients. Ces anomalies biologiques et immunologiques n’ont pas de particularité en fonction de l’âge [15]. La corticothérapie ŕ forte dose (1 mg/kg/j de prednisone) associée aux mesures hygiéno-diététiques usuelles (notamment prévention de l’ostéoporose par les biphosphonates, régime hyposodé, sans sucres rapides et pauvre en graisses saturées), constitue le traitement de premičre intention des M.I. Avant l’utilisation des corticoďdes, le pronostic des DM était médiocre avec une mortalité de 50 ŕ 60% [39]. La DM est corticosensible, monophasique ou plus souvent chronique. Les corticoďdes constituent le traitement de référence et de premičre ligne des myosites. 60 ŕ 70% des DM répondent ŕ la corticothérapie orale ŕ base de prednisone [39]. Le mécanisme d’action du prednisone n’et pas clairement bien établi, mais plusieurs hypothčses ont été avancées. Il pourrait inhiber le recrutement et la migration des lymphocytes dans les différents sites inflammatoires, également interférer avec la synthčse des lymphokines notamment l’interleukine 1, Interleukine 2 et le tumor necrosis factor sécrétées par les macrophages activés et les cellules T [40]. Les corticoďdes sont prescrits ŕ la dose de 1mg/kg/j d’équivalent de prednisone précocement au cours de l’évolution de la maladie. Ces fortes doses sont maintenus pendant 4 ŕ 8 semaines en moyenne, jusqu’ŕ la régression des signes cliniques et la nette diminution ou la normalisation des enzymes musculaires ; une dégression progressive peut ętre par la suite entamée jusqu’ŕ l’obtention de la dose minimale efficace qui sera maintenue pendant 6 ŕ 9 mois sans rechutes [41]. L’utilisation de bolus intraveineux de Méthyl-Prednisolone ŕ la dose de 1g kg j pendant 3j relayé par une corticothérapie forte dose est réservée aux formes sévčres de DM [39]. Dans sa forme paranéoplasique, le traitement de la tumeur peut trčs rarement ętre ŕ lui seul curatif. La corticothérapie est quasiment toujours nécessaire ; la rechute de la DM fait suspecter celle du cancer [2]. Plusieurs facteurs sont associés ŕ une faible réponse au corticoďdes, notamment : le retard ŕ l’instauration des corticoďdes, l’âge avancé, l’atteinte interstitielle pulmonaire et l’atteinte cardiaque, l’association ŕ une néoplasie, la détection d’anticorps de type anti-synthétase et anti-SRP [42]. Pautas et al. n’ont pas trouvé de différence concernant le traitement entre les 2 groupes sujet âgé/sujet jeune, en dehors de la durée du traitement qui était plus longue dans le second groupe (p=0.008) [9]. Les effets secondaires de la corticothérapie étaient plus fréquents chez les sujets âgés. En cas de myosite réfractaire ou corticodépendante, aussi bien pour le sujet jeune que pour le sujet âgé, d’autres options thérapeutiques sont utilisées incluant le methotrexate (MTX), l’azathioprine, le mycophénolate mofetil, les immunoglobulines intraveineuses et le rituximab [9]. On recommande généralement le repos en période de maladie évolutive et d´attendre que les symptômes soient maîtrisés avant de faire de l´exercice pour éviter la perte de tonus musculaire. La prévention des pneumopathies d´inhalation, la kinésithérapie (passive et douce lors des poussées inflammatoires, active et réguličre dčs les premičres semaines de corticothérapie passées, jusqu’ŕ la fin de la corticothérapie) et l´ergothérapie sont indispensables dans la prise en charge de ces patients. La kinésithérapie et la réadaptation musculaire sont absolument indispensables, favorisant le trophisme musculaire, luttant contre la myopathie stéroďdienne et améliorant la vascularisation myocytaire. Lors du retour ŕ domicile aprčs hospitalisation, il est souhaitable que tout soit bien organisé pour recevoir le sujet âgé encore fatigué et souvent sous traitement en évaluant son autonomie et son indépendance aprčs cette pathologie inflammatoire musculaire.
La DM, bien que rare chez la personne âgée, ne doit pas ętre méconnue et doit faire rechercher une pathologie cancéreuse sous-jacente. Notre étude constitue le premier travail réalisé en Tunisie concernant les DM du sujet âgé. Elle nous a permis de mieux connaître les caractéristiques de cette forme et de la comparer aux différentes séries de la littérature. Malgré le traitement intensif par corticoďdes associées parfois ŕ des immunosuppresseurs, ces DM sont grevées d’une importante morbidité et mortalité qui alourdissent le pronostic de la maladie.
Les auteurs ne déclarent aucun conflit d’intéręt.
Tous les auteurs ont contribué ŕ l’élaboration de ce manuscrit. Ils ont lu et approuvé la version finale du manuscrit.
Tableau 1: Comparaison Dermatomyosite du sujet âgé et Dermatomyosite du sujet jeune
Tableau 2: Fréquence des néoplasies associées aux MI chez le sujet âgé selon quelques séries
Tableau 3: Résumé de quelques observations de DM du sujet âgé associée ŕ une néoplasie rapportées dans la littérature
Figure 1: Echographie pelvienne. Masse kystique ovarienne ŕ paroi épaissie renfermant des bourgeonnements tissulaires trčs suspects de malignité
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975; 292:344-347. This article on PubMed
- Dimitri D. Myopathies inflammatoires:diagnostic et classifications. Presse Med. 2009 Jul-Aug;38(7-8):1141-63. This article on PubMed
- Chérin P, Marie I. Les nouveaux critčres diagnostiques et d’évaluation des polymyosites et dermatomyosites. Rev Med Interne. 2005 May;26(5):361-7. This article on PubMed
- Toumi S, Ghnaya H, Braham A, Harrabi I, Laouani-Kechrid C. Les polymyosites et dermatomyosites de l’adulte: Étude multicentrique tunisienne. Rev Med Interne. 2009;30:747–753
- Moulias S. Myopathies du sujet âgé. Revue du Rhumatisme. 2004;71:510 – 514
- Gaubitz M. Epidemiology of connective tissue disorders. Rheumatology (Oxford). 2006 Oct;45 Suppl 3:iii3-4. This article on PubMed
- Vargas-Leguás H, Selva-O'Callaghan A et al. Polymyositis- dermatomyositis: incidence in Spain (1997-2004). Med Clin (Barc). 2007 Nov 24;129(19):721-4. This article on PubMed
- Hengstman GJ, Van Venrooij WJ, Vencovsky J, Moutsopoulos HM, van Engelen BG. The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis. 2000; 59:141-142. This article on PubMed
- Pautas E, Chérin P, Piette JC, Pelletier S, Wechsler B, Cabane J et al. Features of polymyositis and dermatomyositis in the elderly: a case-control study. Clin Exp Rheumatol. 2000;18 :241-244. This article on PubMed
- Marie I, Hatron PY, Levesque H, Hachulla E, Hellot MF, Michon-Pasturel U et al. Influence of age on characteristics of polymyositis and dermatomyositis in adults. Medicine (Baltimore). 1999;78 :139-147. This article on PubMed
- Chen YJ, Wu CY, Huang YL, Wang CB, Shen JL, Chang YT. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010; 12 :R70. This article on PubMed
- Cherin P. Polymyosites et dermatomyosites. Médecine thérapeutique. 2007; 13 :122-134
- Dankó K, Ponyi A, Constantin T et al. Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: A longitudinal study of 162 cases. Medicine (Baltimore). 2004 Jan;83(1):35-42. This article on PubMed
- Lynn SJ, Sawyers SM, Moller PW et al. Adult onset inflammatory myopathy: North Canterbury experience 1989–2001. Intern Med J. 2005 Mar;35(3):170-3. This article on PubMed
- Desnuelle C. Myopathies du sujet âgé. La Lettre du Neurologue. 2007; 11:64
- Machet L, Lavigne C, Rivolllier C. Dematomyosite. Encycl Méd Chir Dermatologie. 2003;98-500-A-10:12
- Koh ET, Seow A, Ong B, Ratnagopal P, Tjia H, Chng HH. Adult onset polymyosistis/drematopolymyositis: clinical and laboratory features and treatment reponse in 75 patients. Ann Rheum Dis. 1993; 52: 857-861. This article on PubMed
- Troyanov Y, Targoff IN, Tremblay JL et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and auto antibodies: anlysis of 100 French Canadian patients. Medicine (Baltimore). 2005 Jul;84(4):231-49. This article on PubMed
- Serratrice G. Dermatopolymyosites. In kahn MF, Peltier AP, Meyer O, Piette J.C. éds. Les maladies systémiques. 1991. Paris:Flammarion Médecine-Sciences
- Ebert EC. Review article: the gastrointestinal complications of myositis. Aliment Pharmacol Ther. 2010; 31: 359-365. This article on PubMed
- Chemtob A, Malka D, Lévy P, Lévy H, Ponsot P, Bernades P. Dysphagie haute et polymyosite. Gastroenterol Clin Biol. 1993;17:502-504. This article on PubMed
- Gabay C. Polyarthrites et connectivites ŕ début tardif. Médecine et hygične. 2001; 59:605-608
- Huang YL, Chen YJ, Lin MW, Wu CY, Liu PC, Chen TJ at al. Malignancies associated with dermatomyositis and polymyositis in Taiwan: a nationwide population-based study. Br J Dermatol. 2009;161:854-860. This article on PubMed
- So MW, Koo BS, Kim YG, Lee CK, Yoo B. Idiopathic Inflammatory Myopathy Associated with Malignancy: A Retrospective Cohort of 151 Korean Patients with Dermatomyositis and Polymyositis. J Rheumatol. 2011 ;38:2432-2435. This article on PubMed
- Sigurgeirsson B, Lindelof B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis - A population-based study. N Engl J Med. 1992; 326: 363-367. This article on PubMed
- Guis S, Mattei JP, Figarella-Branger D, Bendahan D. Myopathies inflammatoires idiopathiques de l’adulte: critčres de diagnostic et de classification. Revue du rhumatisme monographies. 2010;77:99-102
- Hill C, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A et al. Frequency of specific cancer types in dermatomyositis and polymyositis. Lancet. 2001; 357: 96-100. This article on PubMed
- Stockton D, Doherty VR, Brewster DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a scottish population -based cohort study. Br J Cancer. 2001; 85: 41-45. This article on PubMed
- Mebazaa A, Boussen H, Nouira R, Gamoudi A, Rahal K, Kamoun MR at al. Dermatomyosite et cancer du sein: étude Tunisienne multicentrique rétrospective de 13 cas. La Tunisie Medicale. 2011;89 :18-22. This article on PubMed
- Mordel N, Margalioth EJ, Harats N, Ben-Baruch N, Schenker JG. Concurrence of ovarian cancer and dermatomyositis - A report of two cases and literature review. J Reprod Med. 1988 ;33:649-655. This article on PubMed
- Iavazzo C, Vorgias G, Papadakis M, Manikis P, Mavromatis I, Akrivos T. Polymyositis in a patient with recurring ovarian cancer and history of unrelated breast cancer. Arch Gynecol Obstet. 2007;276 :81-84. This article on PubMed
- Cherin P, Piette JC, Herson S, Bletry O, Wechsler B, Frances C et al. Dermatomyositis and ovarian cancer: a report of 7 cases and literature review. J Rheumatol. 1993;20:1897-1899. This article on PubMed
- Wakata N, Kurihara T, Saito E, Kinoshita M. Polymyositis and dermatomyositis associated with malignancy: A 30-year retrospective study. Int J Dermatol. 2002 Nov;41(11):729-34. This article on PubMed
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: A case-control study. Br J Dermatol. 2001 Apr;144(4):825-31. This article on PubMed
- Hu WJ, Chen DL, Min HQ. Study of 45 cases of nasophryngeal carcinoma with dermatomyositis. Am J Clin Oncol. 1996 Feb;19(1):35-8. This article on PubMed
- Fujita Y, Hirano S, Yoneshim Y, Izumi S, Takeda Y, Sugiyama H et al. A case of acute exacerbation of interstitial pneumonia complicated with dermatomyositis during treatment for lung cancer, and literature review. Nihon Kokyuki Gakkai Zasshi. 2011;49:108-115. This article on PubMed
- Vesi? S, Dakovi? Z, Vuki?evi? J, Vukovi? J, Stojkovi? J, Medenica L. Dermatomyositis associated with metastatic rectal adenocarcinoma: case report. Med Pregl. 2009;62:473-475. This article on PubMed
- Kim SW, Shim JS, Eun YG, Kwon KH. Tonsillar squamous cell carcinoma associated with dermatomyositis: the first 2 cases in Korea. Yonsei Med J. 2010;51:605-608. This article on PubMed
- Marie I, Mouthon L. Therapy of polymyositis and dermatomyositis. Autoimmun Rev. 2011;11:6-13. This article on PubMed
- Dalakas MC. Immunotherpy of myositis: issues, concerns and future prospects. Nat Rev Rheumatol. 2010; 6: 129-137. This article on PubMed
- Chérin P. Current therapy for polymyositis and dermatopolymyositis. Rev Med Interne. 2008 Jun;29 Spec No 2:9-14. This article on PubMed
- Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B et al. Polymyositis and dermatomyositis: short term and long term outcome, and predictive factors. J Rheumatol. 2001; 28: 2230-2237. This article on PubMed
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures

Article metrics
Recently from the PAMJ






){kind=link}


Authors´ services
