
Cerebellar ganglioglioma
Badr Fedoul, Zouhayr Souirti
Corresponding author: Fedoul Badr, Neurology Department, Hassan II University hospital, Fez, Morocco 
Received: 28 Feb 2010 - Accepted: 14 Apr 2010 - Published: 22 May 2012
Domain: Clinical medicine
Keywords: Ganglioglioma, cerebellar vermis, histology, tumour, Morocco
©Badr Fedoul et al. Pan African Medical Journal (ISSN: 1937-8688). This is an Open Access article distributed under the terms of the Creative Commons Attribution International 4.0 License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article: Badr Fedoul et al. Cerebellar ganglioglioma. Pan African Medical Journal. 2012;12:12. [doi: 10.11604/pamj.2012.12.12.164]
Available online at: https://www.panafrican-med-journal.com//content/article/12/12/full
Original article 

Cerebellar ganglioglioma
Cerebellar ganglioglioma
Badr Fedoul1,&, Zouhayr Souirti1
1Hassan II University hospital, Fez, Morocco
&Corresponding author
Fedoul Badr, Neurology Department, Hassan II University hospital, Fez, Morocco
Gangliogliomas occur frequently in temporal lobe, and induce incontrollable epilepsy. GG is very rare in cerebellum with 31 cases reported previously [1]. The main elements found in gangliogliomas are atypical neurons, astrocytes, and a fibrovascular stroma. In this communication we report a rare case of GG in the cerebellar hemisphere which was radiologically diagnosed as a pilocystic astrocytoma.
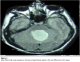
A 27-year-old man complained of diffused headache for 5 months with some episodes of vomiting. There was no significant family history. On neurological examination, there was a static cerebellar syndrome associated with a papilloedema. Brain magnetic resonance (MR) images revealed a cyst lesion in the vermis with ring enhancement after gadolinium administration (Figure 1, Figure 2, Figure 3). The tumor was associated with obstructive hydrocephalus. The cystic portion of tumor showed the cerebrospinal fluid-like signal intensity on T1, T2 MR images but not in FLAIR images (Figure 2). There was no calcification within the tumor on the computed tomography (CT) scan. We performed the operation via a right suboccipital craniectomy. After opening of tensed dura, a cyst was noted and 20 ml of xanthochromic fluid was then aspirated. The wall was totally removed macroscopically. In histological exam, the wall contained atypical neurons and astrocytes and a fibrovascular stroma. Postoperatively, there was a complete recovery. No further treatment was given. At last follow-up, 24 months after surgery; this man had no problems in daily life.
Ganglioglioma is the most commonly encountered glial-neuronal neoplasm of the central nervous system, with its incidence of 0.4% to 4.3% [2]. This tumor is usually seen in children and young adults and there is no gender preponderance [3]. In the series by Chang and associates, of 133 posterior cranial fossa tumors, only 1 case of GG was described. Courville (1930) correctly acknowledged the mixed histological composition of mature ganglion cells and glial cells of varying proportions and degrees of differentiation.
Both components usually exhibit low grades of malignancy, but it is the grade of the glial element that predicts the biological behavior. GG is part of the family of mixed glial ganglion cell tumors that are included within grade I or II of the WHO classification; however, occasional cases of anaplastic GG (WHO grade III) have been reported [4]. Malignant degeneration is also rare, with an estimated incidence of 6%. Although anaplastic transformation of both the glial and neuronal components has been reported, malignant changes appear to be confined mostly to the glial element of the tumor [5] Leptomeningeal and subarachnoid spread is uncommon but may occur [6].
There are no specific clinical findings to indicate cerebellar GG and to discriminate that from other cerebellar lesions. Cerebellar GGs have a generally short mean history (1.6 years) [7] GG may be revealed by static or kinetic cerebellar syndrome and intracranial hypertension syndrome due to obstructive hydrocephalus.
Clinical presentation of infratentorial GG varies depending on the structures involved. Associated symptoms include cranial nerve deficits (hearing loss, intractable facial pain, hemifacial seizures), hemiparesis, gait disturbance, and headache [8]. There are some reports describing epilepsy of cerebellar origin in patients with cerebellar GG. Whether the cerebellar tumor either initiates the seizure or lowers their threshold is still being debated.
Although the exact mechanism for the epilepsy arising in the cerebellum is not known, invasive electrophysiological monitoring with depth electrodes has confirmed the cerebellum as the site of seizure origin. It is thought that the seizure arises in the cerebellum and may generalize secondarily, spreading throughout the cortical surface. Surgical resection of GG has resulted in remission of the seizures in all reviewed cases [9]. Radiologically the cyst with an enhancing mural nodule is classic, but not specific for GGs. The tumor is often isodense or hypodense in the precontrast CT, with calcification present in 6% to 30%. On MR imaging studies, GGs are usually hyperintense on T2-weighted images and isointense to slightly hypointense on T1-weighted images. The variable signal in cystic portion depends on whether the contents are proteinaceous, hemorrhagic, or contain cerebrospinal fluid [2].
Gadolinium enhancement of the solid component of the GGs is observed in about half of the cases, although the pattern varies from intensely homogenous to heterogeneous. Although there is considerable variability in the radiographic appearances of the cerebellar GGs, almost all tumors appear as a discrete, solid/cystic lesion with mild mass effect and little or no surrounding edema [10].
The main differential diagnosis of cerebellar GG in children or young adults is typically the pilocytic astrocytoma [1]. Surgery is the treatment of choice for cerebellar ganglioglioma. Complete resection leads to a good long-term prognosis which results in greater than 90% of 5-year survival. However, because of the location and vicinity to vital and sensitive brain areas and their adherence to the tumor, complete resection of infratentorial GG is often not possible without producing severe deficits or even death, thus leaving only space for subtotal resection [11].
Nevertheless, the diagnosis of GG is of great importance because of its comparatively better prognosis than other tumors in this location. Radiotherapy is not recommended in management of GG because of the entirely radioresistant nature. The radiotherapy doesn?t affect the poetentiel growth of GG [2]. Some reports suggest that postoperative radiation may predispose a GG to malignant degeneration [12].
The cerebellar location of ganglioglioma is exceptional and a diagnostic surprise for the neurosurgereon. It is usually seen in children and young adults. Complete resection is the treatement of choice. Even with partial resection, prognosis remains favorable. Radiotherapy is not required.
The authors declared no competing interests.
Badr Fedoul: Main author providing clinical data, iconography and showing the case interest. Zouhayr Souirti: second author. Discussion. All the authors have read and approve the final version of the manuscript.
Figure 1: Brain MRI T2, axial sequence, showing a hyperintense cyst image in the vermis. The cyst produces a mass effect on brainstem and 4th ventricle
Figure 2: Brain MRI FLAIR, axial sequence, showing a hyperintense signal in the cyst different to CSF signal
Figure 3: Brain MRI T1 sagittal sequence after Gadolinium injection showing homogeneous wall enhancement
- Seong-Ho Park, Ealmaan Kim, Eun-Ik Son. Cerebellar Ganglioglioma. J Korean Neurosurg Soc. 2008; 43: 165-168. This article on PubMed
- Selch Michael, Goy Barry, Lee Steve, El-Sadin Susan, Kincaid Patricia, Park Sun Hye, Withers. Gangliogliomas : experience with 34 patients and review of the literature. Am J Clin Oncol. 1998; 21: 557-564. This article on PubMed
- Rodolfo Hakim, Jay S Loeffler, Douglas C Anthony, Peter M. Black: Gangliogliomas in adults. Cancer. 1997; 79: 127-131. This article on PubMed
- Atushi Sasaki, Junko Hirato, Yoichi Nakazato, Masaru Tamura, Hirotaka Kadowaki. Recurrent anaplastic ganglioglioma: pathological characterization of tumor cells?case report. J Neurosurg. 1996 Jun;84(6):1055-9. This article on PubMed
- Yutaka Hayashi, Masayuki Iwato, Mitsuhiro Hasegawa, Osamu Tachibana, Andreas von Deimling, and Junkoh Yamashita. Malignant transformation of a gangliocytoma/ganglioglioma into a glioblastoma multiforme: a molecular genetic analysis?case report. J Neurosurg. 2001 Jul;95(1):138-42. This article on PubMed
- Robert Tien, Susan Tuori, Nathan Pulkingham, and Peter Burger. Gangliogliomas with leptomeningeal and subarachnoid spread: results of CT, MR, and PET imaging. AJR Am J Roentgenol. 1992 Aug;159(2):391-3. This article on PubMed
- Alfonso Lagares, Pedro A Gómez, Ramiro D Lobato, Jose Ramón Ricoy, Ana Ramos, Adolfo de la Lama. Ganglioglioma of the brainstem: report of three cases and review of the literature. Surg Neurol. 2001 Nov;56(5):315-22; discussion 322-4. This article on PubMed
- am Safavi-Abbasi, Federico Di Rocco, Kraisri Chantra, Guenther C Feigl, Amr El-Shawarby, Amir Samii, and Madjid Samii. Posterior Cranial Fossa Gangliogliomas. Skull Base. 2007 Jul;17(4):253-64. This article on PubMed
- Jong Hee Chae, Seung-Ki Kim, Kyu-Chang Wang, Ki Joong Kim, Yong-Seung Hwang, Byung-Kyu Cho. Hemifacial seizure of cerebellar ganglioglioma origin: seizure control by tumor resection.Epilepsia. 2001 Sep;42(9):1204-7. This article on PubMed
- Josef Zentner, Helmut K Wolf, Burkhard Ostertun, Andreas Hufnagel, Manuel G Campos, Laszlo Solymosi, Johannes Schramm. Gangliogliomas : clinical, radiological and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry. 1994; 57: 1497-1502. This article on PubMed
- Geoffrey Blatt, Arvind Ahuja, Lucia Miller, Peter Ostrow, and Donald Soloniuk. Cerebellomedullary gangliglioma: CT and MR findings. AJNR Am J Neuroradiol. 1995 Apr;16(4):790-2. This article on PubMed
- Rumana Christopher, Valadka Alex. Radiation therapy and malignant degeneration of benign supratentorial gangliogliomas. Neurosurgery. 1998 May;42(5):1038-43. This article on PubMed
Search
This article authors
On Pubmed
On Google Scholar
Citation [Download]
Navigate this article
Similar articles in
Key words
Tables and figures


Article metrics
PlumX Metrics
Cerebellar gangliogliomaRecently from the PAMJ


){kind=link}
){kind=link}
){kind=link}


Authors´ services
